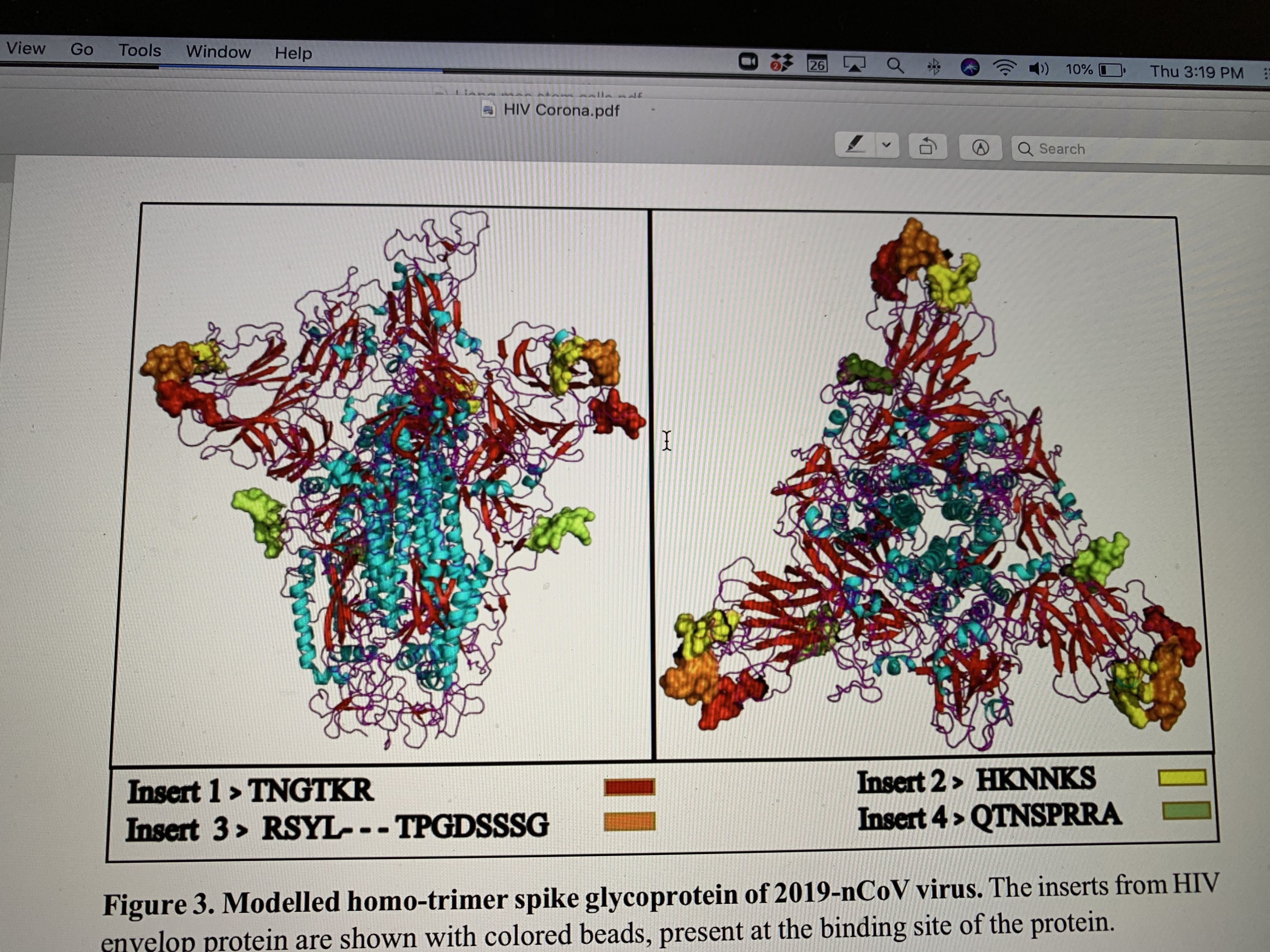
BRAVO EDESTINY (CAPSULES) is a product entirely made in Switzerland and contains exclusively ingredients from certified Swiss and European sources; all the microorganisms, probiotic strains, and ingredients in this product are certified safe for human use. They are manufactured exclusively for our Swiss company by state-of-the-art GMP-certified European facilities that guarantee the highest quality.
In our Swiss facility we choose the best Swiss quality hemp seed protein powder, we blend it with highly selected food ingredients (such as real lemon juice and apple cider vinegar), we add our proprietary blend of fermenting cultures, our array of probiotics and prebiotics, and we leave the product to fully and naturally ferment following our proprietary process. Then, after full and extensive fermentation, we freeze dry the product and we make it available under the form of gastro-resistant (enteric-coated), vegetable, capsules.
I am pleased to announce BRAVO EDESTINY, a completely new product that joins our Bravo family and, just like the other products, it is absolutely unique. Bravo Edestiny is based on fermentation of hemp seed proteins by the proprietary array of probiotic microbes that are at the core of all Bravo products. The two major proteins of hemp are Edestin – hence the name Edestiny – and Albumin.
Edestin has the unique ability to stimulate the body’s immunological responses against invasive agents [1]; fermentation of edestin by Bravo microbes leads to formation of a vegetal analogue of GcMAF and makes Bravo Edestiny the only probiotic with active vegetable based GcMAF. Bravo Edestiny comes in the wake of scientific results demonstrating that it has GcMAF activity 100 fold higher than that of purified GcMAF and lowers nagalase [23].
Albumin, the other protein in Bravo Edestiny, is known to inhibit viral entry into human cells, boost immune responses and attenuate DNA damage [456].
Some of the properties of fermented Edestin and Albumin are mentioned in my latest talk for Sophia Health Institute, [see here] from min 45.
1School of Life Sciences, Shanghai University, Shanghai, 200444, China 2Institute of Basic Medical Sciences Chinese Academy of Medical Sciences, School of Basic Medicine Peking Union Medical College, Beijing, 100005, China 3Beijing You’an Hospital, Capital Medical University, Beijing, China 4Department of Medicine, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China 5Department of Orthopaedics, the First Affiliated Hospital of Zhengzhou University, Zhengzhou, China 6Institute of Stem Cell and Regeneration Medicine, School of Basic Medicine, Qingdao University, Qingdao, Shandong, China 7Department of Orthopaedics, the Second Affiliated Hospital of Xi’an Jiaotong University, Xi’an, China 8Department of Neurosurgery, the First Affiliated Hospital of Zhengzhou University, Zhengzhou, China 9The Executive Committee on Anti-aging and Disease Prevention in the framework of Science and Technology, Pharmacology and Medicine Themes under an Interactive Atlas along the Silk Roads, UNESCO, Paris, France 10International Society on Aging and Disease (ISOAD), Fort Worth, Texas, USA 11The Geriatric Medical Center “Shmuel Harofe”, Beer Yaakov, affiliated to Sackler School of Medicine, Tel-Aviv University, Tel-Aviv, Israel 12University of North Texas Health Science Center, Fort Worth, TX, USA 13School of Biomedical Sciences, Li Ka Shing Faculty of Medicine, University of Hong Kong, Hong Kong, China 14Institute of Chinese Medical Science, University of Macau, Taipa, Macau, China 15Laboratory of Geroprotective and Radioprotective Technologies, Institute of Biology, Komi Science Center of Russian Academy of Sciences, Syktyvkar, Russia 16Department of Pediatrics, Obstetrics and Gynecology, University of Valencia, Valencia, Spain 17Department of Biochemistry, Maharishi Markandeshwar University, Kolkata, India
1
chinaXiv:202002.00080v1
18Institute of Medical Science, University of Toronto, Toronto, Canada 19Department of Biological Sciences, Inha University, Incheon, South Korea 20Centre of Human & Aerospace Physiological Sciences & Centre for Stem Cells and Regenerative Medicine, Faculty of Life Sciences & Medicine, King’s College London, London, UK 21Laboratory of Immunopathology and Immunosenescence, Department of Biomedicine, Neuroscience and Advanced Diagnostics, University of Palermo, Palermo, Italy
#These authors contributed equally to this work
*Corresponding authors: Robert Chunhua Zhao, M.D. & Ph.D., Professor, School of Life Sciences, Shanghai University, Shanghai 200444, China. Email: zhaochunhua@vip.163.com
Kunlin Jin, M.D. & Ph.D., Professor, University of North Texas Health Science Center, Fort Worth, TX, 76107, USA. kunlin.Jin@unthsc.edu
Ronghua Jin, M.D. & Ph.D., Professor, You’an Hospital, Capital Medical University, Beijing, China. Email: jin_eagle@sina.com
Hongjian Liu, M.D. & Ph.D., Professor, Department of Orthopaedics, the First Affiliated Hospital of Zhengzhou University, Zhengzhou, China. Email: hongjianmd@126.com
2
chinaXiv:202002.00080v1
Abstract
A coronavirus (HCoV-19) has caused the novel coronavirus disease (COVID-19) outbreak in Wuhan, China, Preventing and reversing the cytokine storm may be the key to save the patients with severe COVID-19 pneumonia. Mesenchymal stem cells (MSCs) have been shown to possess a comprehensive powerful immunomodulatory function. This study aims to investigate whether MSC transplantation improve the outcome of 7 enrolled patients with COVID-19 pneumonia in Beijing YouAn Hospital, China from Jan 23, 2020. to Feb 16, 2020. The clinical outcomes, as well as changes of inflammatory and immune function levels and adverse effects of 7 enrolled patients were assessed for 14 days after MSC injection. MSCs could cure or significantly improve the functional outcomes of seven patients with COVID-19 pneumonia in 14 days without observed adverse effect. The pulmonary function and symptoms of all patients with COVID-19 pneumonia were significantly improved in 2 days after MSC transplantation. Among them, two common and one severe patient were recovered and discharged in 10 days after treatment. After treatment, the peripheral lymphocytes were increased and the overactivated cytokine-secreting immune cells CXCR3+CD4+ T cells, CXCR3+CD8+ T cells, and CXCR3+ NK cells were disappeared in 3-6 days. And a group of CD14+CD11c+CD11bmid regulatory DC cell population dramatically increased. Meanwhile, the level TNF-α is significantly decreased while IL-10 increased in MSC treatment group compared to the placebo control group. Furthermore, the gene expression profile showed MSCs were ACE2- and TMPRSS2- which indicated MSCs are free from COVID-19 infection. Thus, the intravenous transplantation of MSCs was safe and effective for treatment in patients with COVID-19 pneumonia, especially for the patients in critically severe condition.
The novel coronavirus disease 2019 (COVID-19) has grown to be a global public health emergency since patients were first detected in Wuhan, China, in December 2019. Since then, the number of COVID-19 confirmed patients have sharply increased not only in China, but also worldwide, including Germany, South Korea, Vietnam, Singapore, and USA[1]. Currently, no specific drugs or vaccines are available to cure the patients with COVID-19 infection. Hence, there is a large unmet need for a safe and effective treatment for COVID-19 infected patients, especially the severe cases.
Several reports demonstrated that the first step of the HCoV-19 pathogenesis is that the virus specifically recognizes the angiotensin I converting enzyme 2 receptor (ACE2) by its spike protein[2-4]. ACE2-positive cells are infected by the HCoV-19, like SARS-2003[5,6]. In addition, a research team from Germany revealed that the cellular serine protease TMPRSS2 for HCoV-19 Spike protein priming is also essential for the host cell entry and spread[7], like
3
chinaXiv:202002.00080v1
the other coronavirus (i.e. SARS-2003)[8,9]. Unfortunately, the ACE2 receptor is widely distributed on the human cells surface, especially the alveolar type II cells (AT2) and capillary endothelium[10], and the AT2 cells highly express TMPRSS2[9]. However, in the bone marrow, lymph nodes, thymus, and the spleen, immune cells, such as T and B lymphocytes, and macrophages are consistently negative for ACE2[10]. The findings suggest that immunological therapy may be used to treat the infected patients. However, the immunomodulatory capacity may be not strong enough, if only one or two immune factors were used, as the virus can stimulate a terrible cytokine storm in the lung, such as IL-2, IL-6, IL-7, GSCF, IP10, MCP1, MIP1A, and TNFα, followed by the edema, dysfunction of the air exchange, acute respiratory distress syndrome, acute cardiac injury and the secondary infection[11], which may led to death. Therefore, avoiding the cytokine storm may be the key for the treatment of HCoV-19 infected patients. MSCs, owing to their powerful immunomodulatory ability, may have beneficial effects on preventing or attenuating the cytokine storm.
MSCs have been widely used in cell-based therapy, from basic research to clinical trials[12,13]. Safety and effectiveness have been clearly documented in many clinical trials, especially in the immune-mediated inflammatory diseases, such as graft versus-host disease (GVHD)[14] and systemic lypus erythematosus (SLE)[15]. MSCs play a positive role mainly in two ways, namely immunomodulatory effects and differentiation abilities[16]. MSCs can secrete many types of cytokines by paracrine secretion or make direct interactions with immune cells, leading to immunomodulation[17]. The immunomodulatory effects of MSCs are triggered further by the activation of TLR receptor in MSCs, which is stimulated by pathogen-associated molecules such as LPS or double-stranded RNA from virus[18,19], like the HCoV-19.
Here we conducted an MSC transplantation pilot study to explore their therapeutic potential for HCoV-19 infected patients. In addition, we also explored the underlying mechanisms using a 10× Genomics high throughput RNA sequencing clustering analysis on MSCs and mass cytometry.
Materials and Methods Study design A pilot trial of intravenous MSC transplantation was performed on seven patients with COVID-19 infected pneumonia. The study was conducted in Beijing YouAn Hospital, Capital Medical University, China, and approved by the ethics committee of the hospital (LL-2020- 013-K). The safety and scientific validity of this study “Clinical trials of mesenchymal stem cells for the treatment of pneumonitis caused by novel coronavirus” from Shanghai University/ PUMC have been reviewed by the scientific committee at International Society on Aging and Disease (ISOAD) and issued in Chinese Clinical Trial Registry (ChiCTR2000029990). The Patients The patients were enrolled from Jan 23, 2020 to Jan 31, 2020. All enrolled patients were
4
chinaXiv:202002.00080v1
confirmed by the real-time reverse transcription polymerase chain reaction (RT-PCR) assay of HCoV-19 RNA in Chinese Center for Disease Control and Prevention using the protocol as described previously[11,20]. The sequences were as follows: forward primer 5′-
TCAGAATGCCAATCTCCCCAAC-3′; reverse AAAGGTCCACCCGATACATTGA-3′; andCTAGTTACACTAGCCATCCTTACTGC-3′BHQ1. We initially enrolled patients with COVID-19 (age 18–95 years) according to the guidance of National Health and Health Commission of China (Table 1).
If no improvement signs were observed under the standard treatments, the patient would be suggested to receive the MSC transplantation. Patients were ineligible if they had been diagnosed with any kind of cancers or the doctor declared the situation to belong to the critically severe condition. We excluded patients who were participating in other clinical trials or who have participated in other clinical trials within 3 months.
Cell preparation and transplantation
The clinical grade MSCs were supplied, for free, by Shanghai University, Qingdao Co-orient Watson Biotechnology group co. LTD and the Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences. The cell product has been certified by the National Institutes for Food and Drug Control of China (authorization number: 2004L04792,2006L01037,CXSB1900004). Before the intravenous drip, MSCs were suspended in 100 ml of normal saline, and the total number of transplanted cells was calculated by 1 × 106 cells per kilogram of weight. The window period for cell transplantation was defined as the time when symptoms or/and signs still were getting worse even as the expectant treatments were being conducted. The injection was performed about forty minutes with a speed of ~40 drops per minute.
The patients were assessed by the investigators through the 14-day observation after receiving the investigational product. The clinical, laboratory, and radiological outcomes were recorded and certified by a trained group of doctors. The detailed record included primary safety data (infusional and allergic reactions, secondary infection and life-threatening adverse events) and the primary efficacy data (the level of the cytokines variation, the level of C-reactive protein in plasma and the oxygen saturation). The secondary efficacy outcomes mainly included the total lymphocyte count and subpopulations, the chest CT, the respiratory rate, and the patient symptoms (especially the fever and shortness of breath). In addition, the therapeutic measures (i.e. antiviral medicine and respiratory support) and outcomes were also examined.
Statistical analysis
MIMICS 21.0 (Interactive medical image control system of Materialise, Belgium) was used to evaluate the chest CT data. The analysis of Mass cytometry of the peripheral blood mononuclear cells is described in Supplementary Material 1. The analysis of the 10 x RNA-seq survey is described in Supplementary Material 2. Data were analyzed by SPSS software (SPSS 22.0). Differences between two groups were assessed using unpaired two-tailed t tests. Data
5
primer 5′- the probe 5′CY5-
chinaXiv:202002.00080v1
involving more than two groups were assessed by analysis of variance (ANOVA). P values <0.05 indicated statistical significance.
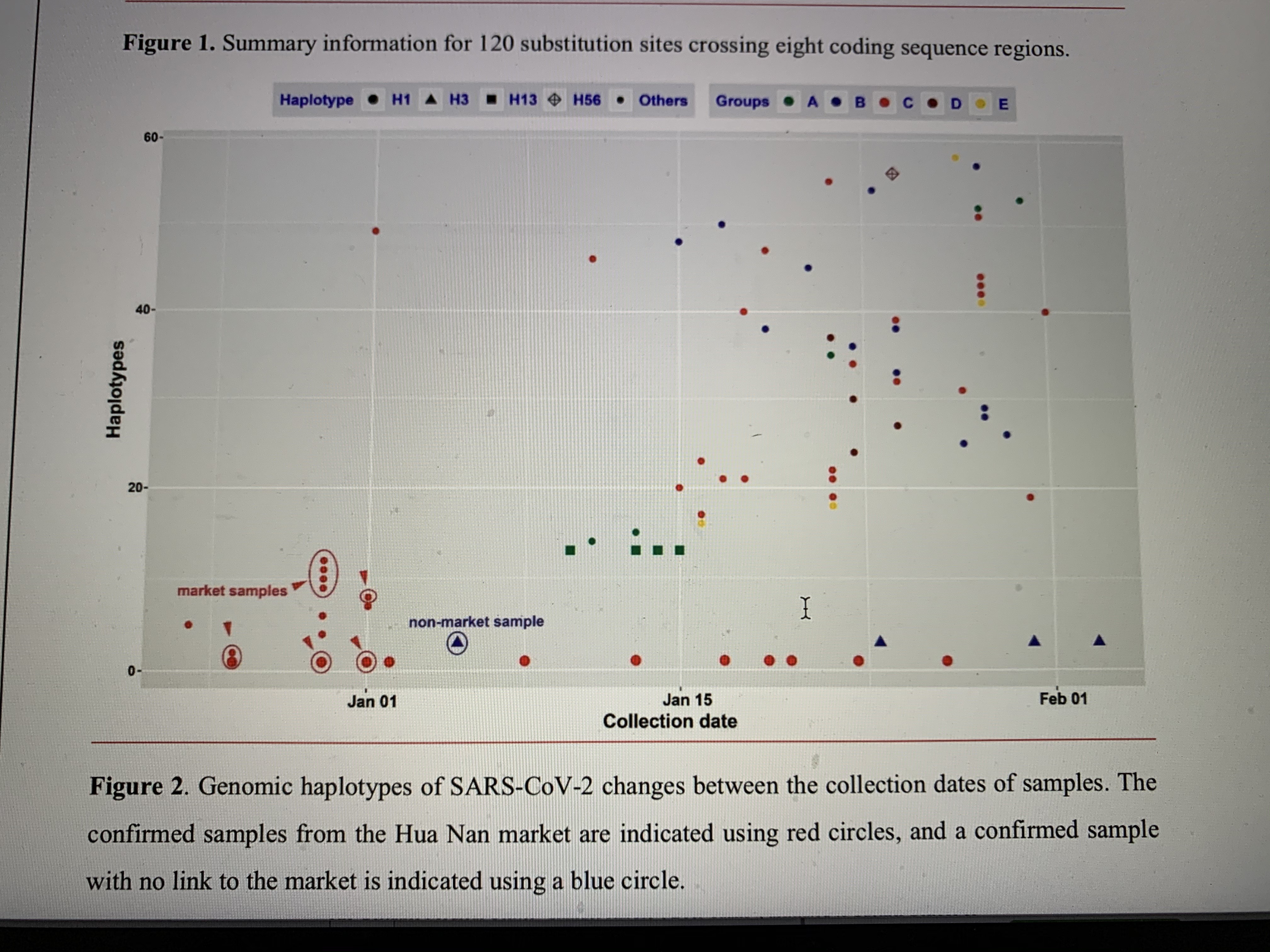
Results MSC treatment procedure and general patient information This study was conducted from Jan 23, 2020, to Feb 16, 2020. Seven confirmed COVID-19 patients, including 1 critically severe type (patient 1), 4 severe types (patient 2, 3, 6, 7) and 2 common types (patient 4, 6) were enrolled. The timepoint of MSC transplantation for each patient is as shown in Figure 1. The general information of the 7 patients is listed in Table 1. Hitherto, the critically severe patient had completed the MSC treatment. This patient had a 10- year medical history of hypertension with the highest-level of 180/90 mmHg recorded. All the treatment information of the patients was collected.
Figure 1. The flow chart of the cell transplantation treatment
The primary safety outcome
Before the MSC transplantation, the patients had symptoms of high fever (38.5°C ± 0.5°C), weakness, shortness of breath, and low oxygen saturation. However, 2~4 days after transplantation, all the symptoms were disappeared in all the patients, the oxygen saturations rose to ≥ 95% at rest, without or with oxygen uptake (5 liters per minute). In addition, no acute infusion-related or allergic reactions were observed within two hours after transplantation. Similarly, no delayed hypersensitivity or secondary infections were detected after treatment. The detailed diagnosis and treatment procedures of the critically severe patient are shown in Supplementary Material 3. The main symptoms and signs are shown in Table 3.
6
chinaXiv:202002.00080v1
The efficacy outcome
The immunomodulating function of MSCs contributed to the main efficacy outcome and the transplantation of MSCs showed impressive positive results (Table 3). For the primary outcome in the critically severe patient 1, the plasma C-reaction protein level decreased from 105.5 g/L (Jan 30) to 10.1 g/L (Feb 13), which reached the highest level of 191.0 g/L on Feb 1, indicating that the inflammation status was alleviating quickly. The oxygen saturation, without Supplementary oxygen, rose from 89% (Jan 31) to 98% (Feb 13), which indicated the pulmonary alveoli regained the air-change function.
The secondary outcomes were also improved (Table 4). Considering, for example, the critically severe patient 1, the lymphopenia was significantly improved after the cell transplantation. The patient was isolated in the hospital isolation ward with a history of hypertension and blood pressure reaching grade 3 hypertension. On Feb 1, biochemical indicators in the blood test showed that aspartic aminotransferase, creatine kinase activity and myoglobin increased sharply to 57 U/L, 513 U/L, and 138 ng/ml, respectively, indicating severe damage to the liver and myocardium. However, the levels of these functional biochemical indicators were decreased to normal reference values in 2~4 days after treatment (Table 4). On February 13, all the indexes reached to normal levels, namely 19 U/L, 40 U/L, and 43 ng/ml, respectively. The respiratory rate was decreased to the normal range on the 4th day after MSC transplantation. Both fever and shortness of breath disappeared on the 4th day after MSCs transplantation. Chest CT imaging showed that the ground-glass opacity and pneumonia infiltration had largely reduced on the 9th day after MSC transplantation (Figure 2).
7
chinaXiv:202002.00080v1
Figure 2. Chest computerized tomography (CT) images of the critically severe COVID-19 patient. On Jan 23, no pneumonia performance was observed. On Jan 30, ground-glass opacity and pneumonia infiltration occurred in multi-lobes of the double sides. Cell transplantation was performed on Jan 31. On Feb 2, the pneumonia invaded all through the whole lung. On Feb 9, the pneumonia infiltration faded away very much. On Feb 15, only little ground-glass opacity was residual in local.
8
chinaXiv:202002.00080v1
HCoV-19 nucleic acid detection
RT-PCR analysis of HCoV-19 nucleic acid was performed before and after MSC transplantation. For the critically severe patient, before transplantation (Jan 23) and 6 days after transplantation (Feb 6), HCoV-19 nucleic acid was positive. 13 days after transplantation (Feb 13), HCoV-19 nucleic acid turned to be negative. The patient 3, 4,5 also turned to be negative result of HCoV- 19 nucleic acid until this report date.
Mass cytometry (CyTOF) analysis of the patients’ peripheral blood
To investigate the profile of the immune system constitution during MSC transplantation, we performed the CyTOF to analyze immune cells in the patients’ peripheral blood before and after transplantation. CyTOF revealed that there was nearly no increase of regulatory T cells (CXCR3-) or dendritic cells (DC, CXCR3-) for the two patients of common type (Patient 4 and 5). But in the severe patients, both the regulatory T cells and DC increased after the cell therapy, especially for the critically severe patient. Notably, no significant CXCR3- DC enhanced after placebo treatment in three severe control patients. Moreover, for the critically severe patient, before the MSC transplantation the percentage of CXCR3+CD4+ T cells, CXCR3+CD8+ T cells, and CXCR3+ NK cells in the patient’s PBMC were remarkably increased compared to the healthy control, which caused the inflammatory cytokine storm. However, 6 days after MSC transplantation, the overactivated T cells and NK cells nearly disappeared and the numbers of the other cell subpopulations were almost restored to the normal levels, especially the CD14+CD11c+CD11bmid regulatory dendritic cell population (Figure 3).
9
chinaXiv:202002.00080v1
Figure 3. The mass cytometry results of peripheral blood mononuclear cells of the enrolled patients (A, B) and the critically severe patient (C). No increase of regulatory T cells (CXCR3-) or dendritic cells (DC, CXCR3-) for the two patients of common type (Patient 4 and 5, Figrue 3A). But in the severe patients, both the regulatory T cells and DC increased after the cell therapy, especially for the critical severe patient 1 (Figure 3B). Moreover, for the critically severe patient 1, before the MSC transplantation the percentage of overactivated CXCR3+CD4+ T cells (#9), CXCR3+CD8+ T cells (#17), and CXCR3+ NK cells (#12) in the patient’s PBMC were remarkably increased compared to the healthy control (Figure 3C). However, 6 days after MSC transplantation, the overactivated T cells and NK cells nearly disappeared and the numbers of the other cell subsets were almost reversed to the normal levels, especially the CD14+CD11c+CD11bmid DC (#20) population. Normal: health individuals, MSCs: mesenchymal stem cells transplant group, Ctrl: placebo control group.
Serum Cytokine/Chemokine/Growth Factor Analysis
After intravenous injection of MSCs, the decrease ratio of pro-inflammatory cytokine in serum TNF-α before and after MSC treatment was significant (p<0.05). Meanwhile, the increase ratio of anti-inflammatory IL-10 (p<0.05) also showed remarkably in the MSC treatment group. The
10
chinaXiv:202002.00080v1
serum levels of chemokines like IP-10 and growth factor VEGF were both increased, though not significantly (Figure 4).
Figure 4. The ratio of serum cytokines IL-10 (A), growth factor VEGF (B), the chemokine IP-10 (C) and TNF-α (D) before and after MSCs treatment were detected in severe patients compared with the control group without MSCs by panel assay analysis, respectively. Ctrl: placebo control group. P-values were determined using the student’s t-test. *P < 0.05.
10 x RNA-seq analysis for transplanted MSCs
To further elucidate the mechanisms underlying MSC-mediated protection for COVID-19 infected patients, we performed the 10 x RNA-seq survey for transplanted MSCs. The 10 x RNA-seq survey captured 12,500 MSCs which were then sequenced with 881,215,280 raw reads totally (Supplementary Material 4). The results revealed that MSCs are ACE2 or TMPRSS2 negative, indicating that MSCs are free from COVID-19 infection. Moreover, anti- inflammatory and trophic factors like TGF-β, HGF, LIF, GAL, NOA1, FGF, VEGF, EGF, BDNF, and NGF were highly expressed in MSCs, further demonstrating the immunomodulatory function of MSCs. Moreover, SPA and SPC were highly expressed in MSCs, indicating that MSCs might differentiate to AT2 cells (Figure 5). KEGG pathway analysis showed that MSCs were closely involved in the antiviral pathways (Supplementary Material 4).
11
chinaXiv:202002.00080v1
Figure 5. The 10 x RNA-seq survey of MSCs genes expression: Both ACE2 (A) and TMPRSS2 (B) were rarely expressed. TGF-β (C), HGF (D), LIF (E), GAL (F), NOA1 (G), FGF (H), VEGF (I), EGF (J), BDNF (K), and NGF (L) were highly expressed, indicating the immunomodulatory function of MSCs. SPA (M) and SPC (N) were highly expressed, indicating MSCs owned the ability to differentiate into the alveolar epithelial cells II. One point represented one cell, and red and gray color showed high expression and low expression,
12
chinaXiv:202002.00080v1
respectively.
Discussion
Both the novel coronavirus and SARS-2003 could enter the host cell by binding the S protein on the viral surface to the ACE2 on the cell surface[3,5]. In addition to the lung, ACE2 is widely expressed in human tissues, including the heart, liver, kidney, and digestive organs[10]. In fact, almost all endothelial cells and smooth muscle cells in organs express ACE2, therefore once the virus enters the blood circulation, it spreads widely. All tissues and organs expressing ACE2 could be the battlefield of novel coronavirus and immune cells. This explains why not only all infected ICU patients are suffering from acute respiratory distress syndrome, but also complications such as acute myocardial injury, arrhythmia, acute kidney injury, shock, and death of multiple organ dysfunction syndrome[11](Figure 6). Moreover, the HCoV-19 is more likely to affect older males with comorbidities and can result in severe and even fatal respiratory diseases such as acute respiratory distress syndrome[21], like the critically severe case here. However, the cure of COVID-2019 is essentially dependent on the patient’s own immune system. When the overactivated immune system kills the virus, it produces a large amount of inflammatory factors, leading to the severe cytokine storms[20]. It suggests that the main reason of these organs damage may be due to virus-induced cytokine storm. Older subjects may be much easier to be affected due to immunosenescence.
Figure 6. ACE2- MSCs benefit the COVID-19 patients via immunoregulatory function
Our 10x scRNA-seq survey shows that MSCs are ACE2- and TMPRSS2- (to the best of our knowledge, it is the first time to be reported) and secrete anti-inflammatory factors to prevent the cytokine storm. They have the natural immunity to the HCoV-19. According to the mass
13
chinaXiv:202002.00080v1
cytometry streaming results, the virus infection caused a total function failure of the lymphocytes, even of the whole immune system. MSCs played the vital immune modulation roles to reverse the lymphocyte subsets mainly through dendritic cells. Our previous study showed that co-culture with MSCs could decrease the differentiation of cDC from human CD34+ cells, while increasing the differentiation of pDC through PGE2[22]. Furthermore, the induction of IL-10–dependent regulatory dendritic cells and IRF8-controlled regulatory dendritic cells from HSC were also reported in rats[23,24]. MSCs could also induce mature dendritic cells into a novel Jagged-2-dependent regulatory dendritic cell population[25]. All these interactions with different dendritic cells led to a shift of the immune system from Th1 toward Th2 responses.
Several reports also focused on lymphopenia and high levels of C-reactive protein in COVID- 19 patients[20,21]. C-reactive protein is a biomarker with high-sensitivity for inflammation and host response to the production of cytokines, particularly TNFα, IL-6, MCP1 and IL-8 secreted by T cells[26]. However, most mechanistic studies suggest that C-reactive protein itself is unlikely to be a target for intervention. C-reactive protein is also a biomarker of myocardial damage[27].
MSC therapy can inhibit the overactivation of the immune system and promote endogenous repair by improving the microenvironment. After entering the human body through intravenous infusion, part of the MSCs accumulate in the lung, which could improve the pulmonary microenvironment, protect alveolar epithelial cells, prevent pulmonary fibrosis and improve lung function.
As reported by Cao’s team[11], the levels of serum IL-2, IL-7, G-SCF, IP10, MCP-1, MIP-1A and TNF-α in ICU patients were higher than those of normal patients. The cytokine release syndrome caused by abnormally activated immune cells deteriorated the patient’s states which may cause disabled function of endothelial cells, the capillary leakage, the mucus block in lung and finally the respiratory failure. And they could cause even an inflammatory cytokine storm lead to multiple organ failure. The administration of intravenous injection of MSCs significantly improved the inflammation situation in severe COVID-19 patients. Due to its unique immunosuppression capacity, the serum levels of pro-inflammatory cytokines and chemokines were reduced dramatically which attracted less mononuclear/macrophages to fragile lung, while induced more regulatory dendric cells to the inflammatory tissue niche. Moreover, the increased IL-10 and VEGF promoted the lung’s repair. Ultimately, the patients with severe COVID-19 pneumonia survived the worst condition and recovery.
Therefore, the fact that transplantation of MSCs improved the outcome of COVID-2019 patients may be through regulating inflammatory response and promoting tissue repair and regeneration.
Acknowledgement
This work was supported by the National Key Research and Development Program of China
14
chinaXiv:202002.00080v1
(2016YFA0101000, 2018YFE0114200), CAMS Innovation Fund for Medical Sciences (2017- I2M-3-007) and the 111 Project (B18007), National Natural Science Foundation of China (81971324, 81672313, 81700782, 81972523, 81771349).
Conflicts of interest
We have no conflicts of interest.
References
[1] Munster VJ, Koopmans M, van Doremalen N, van Riel D, de Wit E (2020). A Novel Coronavirus Emerging in China — Key Questions for Impact Assessment. New England Journal of Medicine.
[2] Xu X, Chen P, Wang J, Feng J, Zhou H, Li X, et al. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. SCIENCE CHINA Life Sciences.
[3] Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, et al. (2020). Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet (London, England).
[4] Zhou P, Yang X-L, Wang X-G, Hu B, Zhang L, Zhang W, et al. (2020). A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature.
[5] Kuba K, Imai Y, Rao SA, Gao H, Guo F, Guan B, et al. (2005). A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nature Medicine, 11:875-879.
[6] Ge X-Y, Li J-L, Yang X-L, Chmura AA, Zhu G, Epstein JH, et al. (2013). Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature, 503:535- +.
[7] Hoffmann M, Kleine-Weber H, Krüger N, Müller M, Drosten C, Pöhlmann S (2020). The novel coronavirus 2019 (2019-nCoV) uses the SARS-coronavirus receptor ACE2 and the cellular protease TMPRSS2 for entry into target cells. bioRxiv:2020.2001.2031.929042.
[8] Glowacka I, Bertram S, Mueller MA, Allen P, Soilleux E, Pfefferle S, et al. (2011). Evidence that TMPRSS2 Activates the Severe Acute Respiratory Syndrome Coronavirus Spike Protein for Membrane Fusion and Reduces Viral Control by the Humoral Immune Response. Journal of Virology, 85:4122-4134.
[9] Iwata-Yoshikawa N, Okamura T, Shimizu Y, Hasegawa H, Takeda M, Nagata N (2019). TMPRSS2 Contributes to Virus Spread and Immunopathology in the Airways of Murine Models after Coronavirus Infection. Journal of Virology, 93.
[10] Hamming I, Timens W, Bulthuis MLC, Lely AT, Navis GJ, van Goor H (2004). Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. Journal of Pathology, 203:631-637.
[11] Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. The Lancet.
[12] Connick P, Kolappan M, Crawley C, Webber DJ, Patani R, Michell AW, et al. (2012). Autologous mesenchymal stem cells for the treatment of secondary progressive multiple sclerosis: an open-label phase 2a proof-of-concept study. Lancet Neurology, 11:150-156.
15
chinaXiv:202002.00080v1
[13] Wilson JG, Liu KD, Zhuo NJ, Caballero L, McMillan M, Fang XH, et al. (2015). Mesenchymal stem (stromal) cells for treatment of ARDS: a phase 1 clinical trial. Lancet Respiratory Medicine, 3:24-32.
[14] Hashmi S, Ahmed M, Murad MH, Litzow MR, Adams RH, Ball LM, et al. (2016). Survival after mesenchymal stromal cell therapy in steroid-refractory acute graft-versus-host disease: systematic review and meta-analysis. Lancet Haematology, 3:E45-E52.
[15] Kamen DL, Nietert PJ, Wang H, Duke T, Cloud C, Robinson A, et al. (2018). CT-04 Safety and efficacy of allogeneic umbilical cord-derived mesenchymal stem cells (MSCs) in patients with systemic lupus erythematosus: results of an open-label phase I study. Lupus Science & Medicine, 5:A46-A47.
[16] Galipeau J, Sensebe L (2018). Mesenchymal Stromal Cells: Clinical Challenges and Therapeutic Opportunities. Cell Stem Cell, 22:824-833.
[17] Bernardo ME, Fibbe WE (2013). Mesenchymal Stromal Cells: Sensors and Switchers of Inflammation. Cell Stem Cell, 13:392-402.
[18] Waterman RS, Tomchuck SL, Henkle SL, Betancourt AM (2010). A new mesenchymal stem cell (MSC) paradigm: polarization into a pro-inflammatory MSC1 or an Immunosuppressive MSC2 phenotype. PLoS One, 5:e10088.
[19] Li W, Ren G, Huang Y, Su J, Han Y, Li J, et al. (2012). Mesenchymal stem cells: a double-edged sword in regulating immune responses. Cell Death Differ, 19:1505-1513.
[20] Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, et al. (2020). Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus–Infected Pneumonia in Wuhan, China. JAMA.
[21] Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, et al. (2020). Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet (London, England).
[22] Chen L, Zhang W, Yue H, Han Q, Chen B, Shi M, et al. (2007). Effects of human mesenchymal stem cells on the differentiation of dendritic cells from CD34(+) cells. Stem Cells and Development, 16:719-731.
[23] Liu X, Qu X, Chen Y, Liao L, Cheng K, Shao C, et al. (2012). Mesenchymal Stem/Stromal Cells Induce the Generation of Novel IL-10-Dependent Regulatory Dendritic Cells by SOCS3 Activation. Journal of Immunology, 189:1182-1192.
[24] Liu X, Ren S, Ge C, Cheng K, Zenke M, Keating A, et al. (2015). Sca-1(+)Lin(-)CD117(-) Mesenchymal Stem/Stromal Cells Induce the Generation of Novel IRF8-Controlled Regulatory Dendritic Cells through Notch-RBP-J Signaling. Journal of Immunology, 194:4298-4308.
[25] Zhang B, Liu R, Shi D, Liu X, Chen Y, Dou X, et al. (2009). Mesenchymal stem cells induce mature dendritic cells into a novel Jagged-2-dependent regulatory dendritic cell population. Blood, 113:46-57.
[26] Sproston NR, Ashworth JJ (2018). Role of C-Reactive Protein at Sites of Inflammation and Infection. Frontiers in Immunology, 9.
[27] Bisoendial RJ, Boekholdt SM, Vergeer M, Stroes ESG, Kastelein JJP (2010). C-reactive protein is a mediator of cardiovascular disease. European Heart Journal, 31:2087-U1505.
16
chinaXiv:202002.00080v1
Table 1: Clinical classification of the COVID-19 released by the National Health and Health Commission of China
Fever, respiratory symptoms, pneumonia performance on X-ray or CT
Meet any of the followings: 1. Respiratory distress, RR ≥ 30/min; 2. Oxygen saturation ≤ 93% at rest state; 3. Arterial partial pressure of oxygen (PaO2) / Fraction of inspiration O2 (FiO2) ≤ 300mnHg, 1mmHg=0.133kPa
Meet any of the followings: 1. Respiratory failure needs mechanical ventilation; 2. Shock; 3. Combined with other organ failure, patients need ICU monitoring and treatment
chinaXiv:202002.00080v1
Table 2: The general information of the enrolled patients.
Patient 1
Patient 2
Patient 3
Patient 4
Patient 5
Patient 6
Patient 7
Ctrl Patient 1
Ctrl Patient 2
Ctrl Patient 3
Gender
M
F
F
F
M
M
M
F
F
F
Age (years)
65
63
65
51
57
45
53
75
74
46
COVID-19 type
Critically severe
Severe
Severe
Common
Common
Severe
Severe
Severe
Severe
Severe
Fever (°C, baseline)
38.6
37.7
38.2
38.5
38.4
39.0
39.0
36.0
38.9
37.7
Shortness of breath
+++
+++
++
+
+
+++
+++
+++
++
+
Oxygen saturation at rest state
89%
93%
92%
95%
94%
92%
90%
91%
92%
93%
Cough, weak, poor appetite
++
+
++
+
++
++
++
+
++
+
Diarrhea
–
–
+
–
–
–
–
–
–
–
Date of diagnosed
Jan 23
Jan 27
Jan 25
Feb 3
Feb 2
Jan 27
Feb 3
Feb 3
Feb 6
Feb 5
Date of intervention (MSCs or Placebo)
Jan 31
Feb 2
Feb 4
Feb 4
Feb 4
Feb 6
Feb 6
Feb 8
Feb 6
Feb 6
Date of recovery
Feb 3
Feb 4
Feb 6 Discharged
Feb 6 Discharged
Feb 5 Discharged
Feb 7
Feb 7
Dead
ARDS
Stable
chinaXiv:202002.00080v1
Table 3: Symptoms, signs and maximum body temperatures of the critically severe patient from Jan 21 to Feb 13, 2020. ICU: Intensive Care Unit; NA: Not Available
Table 4: The laboratory results of the critically severe patient. Red: the value was above the normal. Blue: the value was below the normal. NA: Not Available
Reference range
Jan 24
Jan 30
Jan 31
Feb 1
Feb 2
Feb 4
Feb 6
Feb 10
Feb 13
C-reactive protein (ng/mL)
< 3.00
2.20
105.50
NA
191.00
83.40
13.60
22.70
18.30
10.10
Absolute lymphocyte count (× 109 per liter)
1.10-3.20
0.94
0.60
0.35
0.23
0.35
0.58
0.87
0.73
0.93
White-cell count (× 109 per liter)
3.50-9.50
4.91
6.35
7.90
7.08
12.16
12.57
11.26
10.65
8.90
Absolute neutrophil count (× 109 per liter)
1.80-6.30
3.43
5.43
7.28
6.63
11.33
11.10
9.43
9.18
7.08
Absolute monocyte count (× 109 per liter)
0.10-0.60
0.38
0.25
0.17
0.13
0.35
0.61
0.52
0.48
0.56
Red-cell count (× 1012 per liter)
4.30-5.80
4.69
4.68
4.66
4.78
4.73
4.75
5.16
4.69
4.53
Hemoglobin (g/L)
130.00-175.00
145.00
147.00
145.00
146.00
142.00
145.00
155.00
145.00
137.00
Platelet count (× 109 per liter)
125.00-350.00
153.00
148.00
169.00
230.00
271.00
268.00
279.00
332.00
279.00
Absolute eosinophil count (× 109 per liter)
0.02-0.52
0.02
0.02
0.02
0.02
0.02
0.05
0.15
0.14
0.14
Absolute basophilic count (× 109 per liter)
0.00-0.06
0.02
0.01
0.02
0.02
0.02
0.06
0.10
0.03
0.04
Total bilirubin (μmol/L)
5.00-21.00
7.00
23.00
21.70
19.80
14.20
15.80
16.50
12.50
8.70
Albumin (g/L)
40.00-55.00
41.70
32.30
29.70
29.90
31.60
33.00
32.20
30.10
29.10
Aspartate amino transferase (U/L)
15.00-40.00
14.00
33.00
48.00
57.00
39.00
34.00
23.00
25.00
19.00
Fibrinogen (g/L)
2.00-4.00
2.44
4.24
NA
NA
4.73
NA
3.12
3.84
3.73
Procalcitonin (ng/mL)
< 0.10
0.11
0.12
NA
NA
NA
0.10
0.18
0.15
< 0.10
Creatine kinase isoenzymes (ng/mL)
< 3.60
0.90
0.12
NA
5.67
4.24
NA
0.88
0.90
0.61
Creatine kinase (U/L)
50.00.310.00
168.00
231.00
NA
513.00
316.00
NA
47.00
83.00
40.00
Glomerular filtration rate (ml/min)
> 90.00
81.30
68.00
89.60
99.00
104.00
92.50
108.10
97.10
94.10
Potassium (mmol/L)
3.50.5.30
3.61
2.74
3.00
3.42
3.47
4.18
4.36
4.69
4.61
Sodium (mmol/L)
137.00.147.00
138.50
132.60
129.50
132.80
136.90
135.80
133.80
134.10
137.70
Myoglobin (ng/mL)
16.00.96.00
53.00
80.00
NA
138.00
77.00
NA
62.00
60.00
43.00
Troponin (ng/mL)
< 0.056
0.10
0.07
NA
0.05
0.05
NA
0.02
0.04
0.04
chinaXiv:202002.00080v1
Supplementary Materials: Supplementary 1: The method of Mass Cytometry of peripheral blood mononuclear cells (PBMC) Sample preparation for mass cytometry PBMC samples were collected from COVID-19 infected patients treated with MSCs transplantation at baseline and on Day 6, and PBMC from a healthy donor were set as the control group. All samples were cultured with 2 μM cisplatin (195-Pt, Fluidigm) for 2 minutes before quenching with CSB (Fluidigm) to identify the viability using mass cytometry analysis. A Fix-I buffer (Fluidigm) was then used to fix cells for 15 min at room temperature, followed by washing three times with phosphate buffer solution (PBS). Mass cytometry antibody staining and CD45 barcoding Three samples from the healthy donor, the patient at baseline and Day 6 were stained with CD45 antibodies that were labeled with different metal tags (89, 141 and 172) to minimize internal cross reaction between samples. MaxPar × 8 Polymer Kits (Fluidigm) were used to conjugate with purified antibodies (listed in Supplemental Table 1). All metal-conjugated antibodies were titrated for optimal concentrations before use. Cells were counted and diluted into 1× 106 cells per milliliter in PBS and underwent permeabilization with 80% methanol for 15 minutes at 0°C. After triple washes in CSB, cells were cultured with antibodies in a total 50 μL CSD for 30 min in RT, triple washed in CSB and incubated with 0.125 μm intercalator in fix and perm buffer (Fluidigm) at 4 °C overnight. Data acquisition in Helios After cultured with intercalator, cells were washed three times with ice cold PBS and three times with deionized water. Prior to acquisition, samples were resuspended in deionized water containing 10% EQ 4 Element Beads (Fluidigm) and cell concentrations were adjusted to 1×106 cell/ml. Data acquisition was performed on a Helios mass cytometer (Fluidigm). The original FCS data were normalized and .fcs files for everyone were collected. CyTOF Data Analysis All .fcs files were uploaded into Cytobank, data cleaning and populations of single living cells were exported as .fcs files for further analysis. Files were loaded into R (http://www.rstudio.com), arcsinh transform was performed to signal intensities of all channels. PhenoGraph analysis was performed.
Supplementary 2: The method of the 10 x RNA-seq survey Materials and reagents All supplies and reagents were of the highest grade commercially available. The 0.20 μm-filters, dishes and tubes were purchased from Corning (NY, USA). CD105, CD90, CD44 and CD45 antibodies for the flow cytometry were purchased from Miltenyi Biotec (Bergisch gladbach, Germany). DMEM/F12, fetal bovine serum (FBS), GlutaMAXTM-I, TrypLETM Express, and penicillin and streptomycin antibiotics were purchased from Gibco (California, USA). All other reagents were analytical grade and required no further purification.
17
chinaXiv:202002.00080v1
Supplemental Table 1: Antibodies used in the Mass cytometry analysis.
The mesenchymal stem cells were cultured in DMEM/F12 medium supplemented with 2% FBS, 2%
Antibody Clone
Source
HI30
Fluidigm
HI30
Fluidigm
HIB19
Fluidigm
UCHT2
Fluidigm
NP-6G4
Fluidigm
RPA-T4
Fluidigm
HI100
Biolegend
H1
Fluidigm
RMO52
Fluidigm
NCAM16.2
Fluidigm
Bu15
Biolegend
3G8
Biolegend
MAb11
Fluidigm
DREG-56
Fluidigm
Polyclonal
Abcam
L128
Fluidigm
G025H7
Fluidigm
B27
Fluidigm
G043H7
Fluidigm
CD28.2
Fluidigm
BC96
biolegend
RPA-T8
Fluidigm
TW46H10
Fluidigm
UCHL1
Fluidigm
Polyclonal
Abcam
JES3-9D7
Fluidigm
MQ2-13A5
Biolegend
15-2
Fluidigm
ML5
Fluidigm
UCHT1
Fluidigm
Y1/82A
Fluidigm
HI30
biolegend
L243
Fluidigm
MP4-25D2
Biolegend
A019D5
Fluidigm
ICRF44
Fluidigm
18
chinaXiv:202002.00080v1
GlutaMAXTM-I, 1% antibiotics and 2 mM GlutaMAXTM-I at 37°C with 5% CO2. After three passages, MSCs were immune-phenotyped by flow cytometry for the following surface markers: CD105, CD90, CD73, CD29, HLA-DR, CD44, CD14 and CD45 (all antibodies from BD Pharmingen, San Jose, USA). And MSCs were tested for adipogenic, chondrogenic and osteogenic differentiation to identify their characters.
Cell preparation and Library construction
Cell count and viability were examined by microscope after 0.4% trypan blue coloring. When the viability was no lower than 80%, the library construction was performed. Library was constructed using the Chromium controller (10 x Genomics, Pleasanton, CA). Briefly, single cells, reagents and Gel Beads containing barcoded oligonucleotides were encapsulated into nanoliter-sized GEMs (Gel Bead in Emulsion) using the GemCode technology. Lysis and barcoded reverse transcription of polyadenylated mRNA from single cells were performed inside every GEM. Post RT-GEMs were cleaned up and cDNA were amplified. cDNA was fragmented and fragment ends were repaired, as well as A-tailing was added to the 3’ end. The adaptors were ligated to fragments which were double sided SPRI selected. Another double sided SPRI selecting was carried out after sample index PCR. Quality control-pass libraries were sequenced. The final library was quantitated in two ways: determining the average molecule length using the Agilent 2100 bioanalyzer instrument; and quantifying the library by real-time quantitative PCR.
Analysis of single-cell transcriptomics data
The reads were demultiplexed by using the Cell Ranger Single Cell Software Suite (v3.1.0, 10 x Genomics) and R package Seurat (v3.1.0). The number of genes, unique molecule identifier (UMI) counts and percentage of mitochondrial genes were examined to identify outliers. Principal component analysis was used for dimensionality reduction. U-MAP was then used for two- dimensional visualization of the results. DEGs were identified with the FindConservedMarkers function in Seurat by parameters of logfc.threshold >0.25, minPct>0.25 and Padj≤0.05. KEGG pathways with FDR ≤0.05 were considered to be significantly enriched.
Supplementary 3: The detailed diagnosis and treatment procedures for the critically severe patient On the evening of January 22, 2020, a 65-year-old man presented to the emergency department of Beijing YouAn Hospital, Beijing, with a 2-day history of cough, sputum and subjective fever. The patient wore a mask in the hospital. He disclosed to the physician that he had traveled in Wuhan, China, from December 31, 2019 to January 20, 2020 and returned to Beijing on January 20. Apart from a 10-year history of hypertension with the highest blood pressure of 180/90 mmHg ever, the patient had no other specific medical history. The physical examination showed a body temperature of 37.8, blood pressure of 138/85 mmHg, pulse of 85 beats per minute, respiratory rate of 19 breaths per minute. Lung auscultation revealed rhonchi. A blood routine examination was arranged urgently, and the result revealed that the white-cell count and absolute lymphocyte count were 4.9 × 109/L (reference range (3.5~9.5) × 109/L) and 0.94 × 109/L (reference range (1.1~3.2) × 109/L),
19
chinaXiv:202002.00080v1
respectively (Table 1). According to the COVID-19 guidance released by the National Health Commission of China, the physician gave him a diagnosis of a suspected COVID-19 case and asked him to undergo medical isolation observation in the hospital. Meantime, the doctor collected his oropharyngeal swab specimen.
On January 23, 2020, the RT-PCR assay confirmed that the patient’s specimen tested positive for HCoV-19. Then the patient was admitted to an airborne-isolation unit in Beijing YouAn Hospital for clinical observation. He had no dyspnea. His consciousness was clear, and the diet and sleep were normal since he became sick. A chest computed tomography (CT) was reported as showing no evidence of infiltrates or abnormalities. The admitting diagnoses were new coronary pneumonia (common type) and hypertension III. The patient received no special care except the irbesartan, which was taken all through the treatment period.
On January 24 to January 29, the patient’s vital physical signs remained largely stable, apart from the development of intermittent fevers and shortness of breath. During this time, the patient received antipyretic therapy including 15 ml of ibuprofen suspension every 6 hours and 650 mg of acetaminophen every 6 hours. From January 26, the patient also received antiviral therapy including lopinavir and ritonavir twice a day, with the amount of 400 mg and 100 mg each time, respectively. On January 30, the patient felt severe shortness of breath and appeared fatigued. The oxygen saturation values measured by pulse oximetry decreased to as low as 91% while he was breathing ambient air. Auscultation rhonchi became worse in the middle of the double sides of the lung. An urgent chest CT clearly showed evidence of pneumonia, ground-glass opacity, in the middle lobes of the right and left lung. The other positive results of laboratory tests included the C-reactive protein rise to 105.5 g/L (reference range < 3 g/L), but the absolute lymphocyte count decreased to 0.60 × 109/L. The potassium concentration went down to 2.74 mmol/L (reference range 3.5-5.5 mmol/L). The doctors decided to change the diagnosis to COVID-19 (critically severe type), and the patient was admitted to ICU unit. More treatments were conducted consisting of mask oxygen supplementation (5 liters per minute), electrocardiograph monitoring, potassium chloride sustained release tablets (oral, 500 mg per time, 3 times per day) and more glucose and amino acid injection. Finally, the discomfort was released, and the oxygen saturation increased to 95%.
On January 31, the shortness of breath even got worse under the oxygen supplementation. The doctor speeded up the oxygen airflow to 10 liters per minute. After the patient signed an agreement to perform the MSCs transplantation, 100 ml of normal saline including 6 × 107 MSCs was intravenously injected into the patient, and no adverse events were observed in association with the infusion.
On February 1 and 2, the patient did not feel better. The third chest CT revealed that the pneumonia got worse. On February 1, the levels of C-reactive protein were 191.0 g/L, and the absolute lymphocyte count decreased badly to 0.23 × 109/L. The laboratory results showed that his liver and myocardium were very likely to be affected. The electrocardiograph monitoring showed the blood pressure, heart rate, respiratory rate and oxygen saturation were 138/80 mmHg, 95 bpm, 33 bpm and 93% under the mask oxygen supplementation of 10 liters per minute. The doctors informed the
20
chinaXiv:202002.00080v1
patient’s families of a critical condition. However, the patient felt better on February 3, for instance, the shortness of breath was significantly recovering. On February 4, the C-reactive protein decreased to 13.6 g/L, and the absolute lymphocyte count rose to 0.58 × 109/L, which indicated that the patient was recovering rapidly. The indexes of liver and myocardium function recovered. Both fever and shortness of breath disappeared on February 5. He was rolled out of ICU. On February 9, the fourth chest CT confirmed that the pneumonia was disappearing. On February 13, the C-reactive protein concentration was 10.1 g/L, and the absolute lymphocyte count was 0.93 × 109/L. Up to now, the patient felt much better.
Supplementary 4: More results of the 10 x RNA-seq surevey Flow cytometry analysis The PI staining results showed that 91.60% of the total cell population was alive, and the cells were: CD105+, CD90+, CD73+, CD44+, CD29+, CD14- and CD45- (Supplemental Figure 1).
Supplemental Figure 1. Flow cytometry evaluation of transplanted MSCs (A) Single cells (87%) were gated firstly. (B) Live cells (91% of the single cells) were enrolled. (C-F) 99% of selected cells were CD105+, CD90+, CD73+, CD44+, CD29+, CD14- and CD45-.
The overview of the survey
A deep transcriptional states map of MSCs and gene expression at single-cell level was generated after the performance of 10× Genomics high throughput of RNA sequencing. The 12,500 cells were acquired in the survey, leading to 881,215,280 raw reads totally. The median number of genes and UMIs detected per cell were 4,099 and 23,971, respectively (Supplemental Figure 2). The sequencing saturation rate was 72.9%, which met the scRNA-seq requirements.
21
chinaXiv:202002.00080v1
Supplemental Figure 2. In the 10 x RNA-seq survey, the median number of genes and UMIs detected per cell were 4,099 (A) and 23,971 (B) as showed in the violin distribution.
MSCs marker genes expression
The scRNA-seq showed that the MSCs highly expressed ENG (CD105), THY1 (CD90), and NT5E (CD73). However, the expression of PTPRC (CD45), CD34, CD14, CD19, and HLA-DR was nearly undetected in the cells (CD45 was the only one shown in Supplemental Figure 3). The results were in accordance with the flow cytometry analysis. In Supplemental Figure 3, one point meant one cell, and red and gray color represented high expression and low expression, respectively.
Supplemental Figure 3. MSCs marker genes expression by 10 x scRNA-seq analysis. (A) CD105+, (B) CD90+, (C) CD73+, and (D) CD45-
ACE2 gene expression and DEGs between ACE2+ MSC and ACE2– MSC
Only one of the 12,500 cells was ACE2+ as shown in Supplemental Figure 4A. Furthermore, the top 60 DEGs between the ACE2+ MSC and the other nearby ACE2- MSC were shown in Supplemental Figure 4B. It is revealed that the ACE2+ MSC tended to generate pro-inflammatory function by
22
chinaXiv:202002.00080v1
secreting IL-8, IL-6 and so on, while ACE2- MSC tended to generate anti-inflammatory effect by secreting BDNF and other factors.
Supplemental Figure 4. (A) ACE2 gene expression in MSCs. (B) top 60 DEGs between one ACE2+ MSC and one ACE2- MSC
23
chinaXiv:202002.00080v1
TMPRSS2 gene expression and DEGs between TMPRSS2 + MSC and TMPRSS2 – MSC
Only seven of the 12,500 cells were TMPRSS2+ as shown in Supplemental Figure 5A. Furthermore, the top 60 DEGs between the TMPRSS2+ MSC and the other seven nearby TMPRSS2- MSC were shown in Supplemental Figure 5B.
Supplemental Figure 5. (A) TMPRSS2 gene expression in MSCs. (B) top 60 DEGs between seven TMPRSS2+ MSCs and seven TMPRSS2- MSCs
24
chinaXiv:202002.00080v1
Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis
KEGG pathway analysis demonstrated diseases mainly related to viral infectious diseases, cancers and endocrine and metabolic disorders (1727 genes, 1605 genes and 1384 genes, respectively). Organismal systems mainly related to endocrine and immune systems (1578 genes and 748 genes, respectively) (Supplemental Figure 6). Four enriched KEGG pathways were also involved in viral infection (Supplemental Figure 7).
Supplemental Figure 6. Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis revealed that many gene expressions of MSCs were related with endocrine and immune systems.
25
chinaXiv:202002.00080v1
Supplemental Figure 7. Four enriched KEGG pathways were also involved in viral infection.
Furin, a potential therapeutic target for COVID-19
Canrong WU,a,1Yueying YANG,b,1Yang LIU,bPeng ZHANG,bYaliWANG,bQiqi WANG, b Yang XU,bMingxue LI,bMengzhu ZHENG,a,* Lixia CHEN,b,* &Hua LIa,b,*
aHubei Key Laboratory of Natural Medicinal Chemistry and Resource Evaluation, School of Pharmacy, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, China bWuya College of Innovation, Key Laboratory of Structure-Based Drug Design & Discovery, Ministry of Education, Shenyang Pharmaceutical University, Shenyang 110016, China
1 These authors contributed equally to this work. *Corresponding author: Hua Li (E-mail: li_hua@hust.edu.cn).
A novel coronavirus (SARS-CoV-2) infectious disease has broken out in Wuhan, Hubei Province since December 2019, and spread rapidly from Wuhan to other areas, which has been listed as an international concerning public health emergency. We compared the Spike proteins from four sources, SARS-CoV-2, SARS-CoV, MERS-CoV and Bat-CoVRaTG13, and found that the SARS-CoV-2 virus sequence had redundant PRRA sequences. Through a series of analyses, we propose the reason why SARS-CoV-2is more infectious than other coronaviruses. And through structure based virtual ligand screening, we foundpotentialfurin inhibitors, which might be used in the treatment of new coronary pneumonia.
In December 2019, a series of acute respiratory diseases occurred in Wuhan, Hubei Province, China and then spread rapidly from Wuhan to other areas. As of February 17, 2020, a total of 71,444 patients have been diagnosed and 1,775 have died worldwide. This is caused by a novel coronavirus, which was named as “2019-nCoV” by the World Health Organization, and diseases caused by 2019-nCoV was COVID-19. 2019-nCoV, as a close relative of SARS-CoV, was classified as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) by the International Committee on Taxonomy of Viruses (ICTV) on February 11, 2020.
Coronaviruses (CoVs) are mainly composed of four structural proteins, including Spike (S), membrane (M), envelope (E) and nucleocapsid (N) [1]. Spike, a trimeric glycoprotein of CoVs, determines diversity of CoVs and host tropism, and mediates CoVs binding to host cells surface-specific receptors and virus-cell membrane fusion [2]. Current research found that SARS-CoV-2 belongs to the beta coronavirus genus, and speculated that it may interact with angiotensin-converting enzyme 2 (ACE2) on the surface of human cells through Spike protein, thereby infecting human respiratory epithelium cell [3]. Letko M and Munster Vthen identified the receptor for SARS-CoV-2 entry into human cells to be ACE2 [4].
Coronavirus Spike protein plays a key role in the early stages of viral infection, with the S1 domain responsible for receptor binding and the S2 domain mediating membrane fusion [5]. The process of SARS-CoV infecting the host involves two indispensable cleaving processes which affect the infectious capacity of SARS-CoV. First, Spike was cleaved into receptor-bound N-terminal S1 subunit and membrane-fusion C-terminal S2 subunit by host proteases at S1/S2 cleavage site (such as type II transmembrane serine protease (TMPRSS2), cathepsins B and L) [6,7]. Second, after CoVs are endocytosed by the host, the lysosomal protease mediates cleavage of S2 subunit (S2’ cleavage site) and releases the hydrophobic fusion peptide to fuse with the host cell membrane [8].
Furin, a kind of proprotein convertases (PCs), is located in the trans-Golgi network (TGN) and activated by acid pH [9]. Furin can cleave precursor proteins with specific motifs to produce mature proteins with biological activity. The first (P1) and fourth (P4) amino acids at the N-terminus of the substrate cleavage site must be arginine “Arg-X-X-Arg ↓” (R-X-X-R,X: any amino acid, ↓:cleavage site). If the P2 position is basic lysine or arginine, the cleavage efficiency
chinaXiv:202002.00062v1
can be improved by about 10 times [10]. Kibler KV et al. demonstrated that the Spike protein S1/S2 and S2′ cleavage sites of the infectious bronchitis virus (IBVs) Beaudette strain can be recognized by fruin, which is a distinctive feature of IBV-Beaudette with other IBVs and has stronger infection ability [11,12]. Based on the characteristics of furin’s recognition substrate sequence, some short peptide inhibitors have been developed, such as Decanoyl-Arg-Val-Lys-Arg-chloromethylketone (Dec-RVKR-CMK) and modified α1-antitrypsin Portland (α1-PDX). However, the non-specific and irreversible inhibitory effects on all members of the PC family limit their application [10, 13]. No small molecule inhibitor of furin with good effect and high specificity has been found so far.
The epidemiological observations showed the infectious capacity of SARS-CoV-2 is stronger than SARS-CoV, so there are likely to be other mechanisms to make the infection of SARS-CoV-2 easier. We suppose the main possibilities as follows, first, SARS-CoV-2 RBD combining with ACE2 may have other conformations; second, the SARS-CoV-2 Spike protein can also bind to other receptors besides ACE2; third, Spike is more easily cleaved by host enzymes and easily fuses with host cell membrane. We compared the Spike proteins from four sources, SARS-CoV-2, SARS-CoV, MERS-CoV and Bat-CoVRaTG13, and found that the SARS-CoV-2 virus sequence had redundant PRRA sequences. Through a series of analyses, this study propose that one of the important reasons for the high infectivity of SARS-CoV-2 is a redundant furin cut site in its Spike protein.And through structure based virtual ligand screening, we proposed possible furin inhibitors, which might be potentially used in the treatment of COVID-19.
2. Methodology 2.1 Homology Spike protein blast and sequence alignment.
The Spike protein of(GB:QHR63250.1) was downloaded from NCBI nucleotide database. The protein sequence were aligned with whole database using BLASTp to search for homology viral Spike protein (Alogorithm parameters, Max target sequences: 1000, Expect threshold: 10). Multiple-sequence alignment was conducted in BLASTp online and analysis with DNAMAN and Jalview. The evolutionary history was inferred using the Neighbor-Joining method in MEGA 7 software package. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test wasdetermined by 500 replicates. The Spike protein sequence analyses were
chinaXiv:202002.00062v1
conducted in snapgene view.
2.2 Furin cleavage site prediction
The prediction of furin cleavage sites were carried out in ProP 1.0 Server (http://www.cbs.dtu.dk/services/ProP/). 2.3 Compounds database
Approved drug database was from the subset of ZINC database, ZDD (ZINC drug database) containing 2924 compounds [14]. Natural products database was constructed by ourselves, containing 1066 chemicals separated from traditional Chinese herbals in own lab and natural-occurring potential antiviral components and derivatives. Antiviral compounds library contains 78 known antiviral drugs and reported antiviral compounds through literature search.
2.4 Homology modeling and molecular docking
Corresponding homology models predicted by Fold and Function Assignment System server for each target protein were downloaded from Protein Data Bank (www.rcsb.org). Alignment of two protein sequences and subsequent homology modeling were performed by bioinformatics module of ICM 3.7.3 modeling software on an Intel i7 4960 processor (MolSoft LLC, San Diego, CA). For the structure-based virtual screening, ligands were continuously resiliently made to dock with the targetthat was represented in potential energy maps by ICM 3.7.3 software, to identify possible drug candidates. 3D compounds of each database were scored according to the internal coordinate mechanics (Internal Coordinate Mechanics, ICM)[15]. Based on Monte Carlo method, stochastic global optimization procedure and pseudo-Brownian positional/torsional steps, the position of intrinsic molecular was optimized. By visually inspecting, compounds outside the active site, as well as those weakly fitting to the active site were eliminated. Compounds with Scores less than -30 or mfScores less than -100 (generally represents strong interactions) have priority to be selected. Protein-protein docking procedure was performed according to the ICM-Pro manual.
3. Results 3.1 Bioinformatics analysis reveals furin cut site in Spike protein of SARS-CoV-2
By sequence alignment of Spike protein sequence of SARS-CoV-2 with its highly homologous sequences, it was found that cleavage site Spike of SARS-CoV-2 had 4 redundant
chinaXiv:202002.00062v1
amino acids-PRRA, and these were not found in those of high homology coronavirus, which formed a furin-like restriction site as RRAR(Figure S1). Through prediction in ProP 1.0 Server, it was found the sequence was indeed easily digested by furin(Figure S2). In order to explore the evolution of this sequence, we used the BLASTp method to find 1,000 homologous Spike sequences with homology from 100% to 31%, which all from beta CoVs. Multiple sequence alignments were performed on these thousands of Spike sequences. One sequence was selected from each highly homologous class (homology greater than 98.5%) for further sequence alignment, and about 155 sequences were finally selected. A homologous multiple sequence alignment was performed on these 155 sequences, and then a phylogenetic tree was constructed(Figure 1). It is found from the phylogenetic tree that the Spike of SARS-CoV-2 exhibited the closest linkage to those of Bat-SL-CoV and SARS-CoV, and far from those of MERS-CoV, HCoV-HKU1, HCoV-OC43. In general, most of the Spike protein in α-CoV does not have a furin cleavage site, most of that in gama-CoV has a furin cleavage site, and that in beta-CoV with or without furin cleavage site are common[16].
We performed furin digestion site prediction on the sequence of each type of coronavirus Spikethrough online software. It was found that all Spike with a SARS-CoV-2 Spike sequence homology greater than 40% did not have a furin cleavage site (Figure 1, Table 1), including Bat-CoV RaTG13 and SARS-CoV (with sequence identity as 97.4% and 78.6%, respectively). The furin cleavage site “RRAR” in SARS-CoV-2 is unique in its family, rendering by its unique insert of “PRRA”. The furin cleavage site of SARS-CoV-2 is unlikely to have evolved from MERS, HCoV-HKU1, and so on. From the currently available sequences in databases, it is difficult for us to find the source. Perhaps there are still many evolutionary intermediate sequences waiting to be discovered.
By analysis of the SARS -CoV-2 Spike protein sequence, it was found that most features are similar to SARS-CoV. It has an N-terminal signal peptide and is divided into two parts, S1 and S2. Among them, S1 contains N-terminal domain and receptor binding region. And S2 is mainly responsible for membrane fusion. The C-terminal region of S2 is S2′, containing a fusion peptide, Hetad repeat1, Hetad repeat 2, and a transmembrane domain(Figure 2). There are two cleavage sites between S1 and S2 ‘, named CS1 and CS2. However, there are some differences in this two cleavage sites.
chinaXiv:202002.00062v1
Unlike SARS-CoV, SARS-CoV-2 contains polybasic amino acids (RRAR) at the CS1 digestion site, and trypsin digestion efficiency will be significantly improved here[5]. More importantly, as mentioned above, this site can be recognized and cleaved by the furin enzyme. The cleavage of Spike protein promotes structural rearrangements of RBD for the adaptation to receptor, thus increasing the affinity[17]. More importantly, the digestion of Spike is an indispensable for membrane fusion of S2 part[18]. In this case, the efficiency of the SARS-CoV-2Spike protein cleavage is significantly higher than that of SARS-CoV, and the SARS-CoV-2Spike protein could be cut during the process of virus maturation (Figure 3). The receptor affinity and membrane fusion efficiency of SARS-CoV-2 would be significantly enhanced compared to that of SARS-CoV. The membrane fusion of SARS-CoV-2Spike protein is more likely to occur during endocytosis process. This may explains the current strong infectious capacity of SARS-CoV-2. So, the development of furin inhibitors may be a promising approach to block its transmissibility.
Figure1.Evolutionary relationships of taxa.The evolutionary history was inferred using the Neighbor-Joining method. The bootstrap consensus tree inferred from 500 replicates is taken to represent the evolutionary history of the taxa analyzed. Branches corresponding to partitions reproduced in less than 50% bootstrap replicates are collapsed. The evolutionary distances were
chinaXiv:202002.00062v1
computed using the Poisson correction method and in the units of the number of amino acid substitutions per site. The analysis involved 155 amino acid sequences. All positions containing gaps and missing data were eliminated. There are a total of 711 positions in the final dataset. Evolutionary analyses were conducted in MEGA7. Those painted in red mean containing cleavage site in sequences and those painted in yellow mean no cleavage site in sequences.
Figure 2.Sequence analysis of Spike protein in SARS-CoV-2. It contains an N-terminal signal peptide, S1 and S2. S1 contains N-terminal domain and receptor binding region. And S2 is mainly responsible for membrane fusion. The C-terminal region of S2 is S2′, it contains a fusion peptide, HR1, HR2, and a transmembrane domain, the amino acid sequence numbers of every domain are annotated below them. Cleavage sites contained in SARS-CoV and SARS-CoV-2 are marked by rhombus.
Figure 3.A schematic diagram of the process of SARS-CoV and SARS-CoV-2 infecting host cells.Those protease are presented by sector in different colors. Furin can cleaveSpike in the process of viral maturation.
chinaXiv:202002.00062v1
Table 1.Furin cleavage probability of Spike sequence homology
aScores are predicted by ProP 1.0 Server. Scores above 0.5 mean furin cleavable. bIdentities compared with SARS-CoV-2 Spike protein.
3.2 Homology modeling and protein-protein docking calculation
In our previous studies (accepted by ActaPharmaceuticaSinica B), both SARS and SARS-CoV-2 spike RBD structures have been docked with human ACE2 to calculate their binding free energy. In that time, the complex structure of SARS-CoV-2 RBD with ACE2 was not available.Its energy was calculated based on the homology model generated from
chinaXiv:202002.00062v1
SARS_RBD-ACE2 complex. The binding energy between the SARS-CoV-2 spike RBD and human ACE2 was -33.72 kJ mol-1, and that between SARS-CoV spike RBD and ACE2 was -49.22 KJ mol-1.This means the binding affinity between SARS-CoV-2 spike and ACE2 is weaker than that of SARS spike. During this manuscript was prepared, the structure of SARS-CoV-2 spike RBD-ACE2 complex was disclosed[19]. Based on this new real structure of SARS-CoV-2 spike RBD-ACE2 complex, we re-did the calculation and found that the binding free energy between SARS-CoV-2 spike RBD and ACE2 was -50.13 KJ mol-1 (Figure S3). This means the binding affinity between SARS-CoV-2 spike and ACE2 is slightly stronger than that of SARS spike.By inspecting the crystal structure of SARS-CoV-2 RBD-ACE2 complex and SARS RBD-ACE2 complex, one can find that one key loop of SARS-CoV-2 RBD in the complex interface had very different conformation compared to that of SARS RBD and previous modeled SARS-CoV-2 RBD (Figure S4).
In order to further explore the possible mechanism how furin cleaves SARS-CoV-2 Spike, we perform protein-protein docking for furin and Spike. Although a Cryo-EM structure of SARS-CoV-2 Spike has been published in bioRxiv during this manuscript was prepared[20], the PDB coordinate was still not available so far. We already built a homology model of SARS-CoV-2 Spike in our previous paper submitted to another regular journal. SARS-CoV-2 Spike structure was built by using the SARS-CoVSpike structure as the temple (PDB code: 5X58)[21]. By superimposing the SARS-CoVSpike with the SARS-CoV-2 Spike, we can find that the major conformation differences between two structures are RBD domain, Arg685/677 loop region(furin/trypsin/TMPRSS2 cut site) and S2 loop region just after fusion peptide (Figure 4A).The trypsin/TMPRSS2 cut site of SARS-CoV was disordered and missing from the original Cryo-EM structure possibly due to its flexibility and without electro density. The “PRRA” inserting in SARS-CoV-2 in this region apparently generate the more flexible loop region and accessible cut site for protease. We performed protein-protein docking by setting SARS-CoV-2 Spikefurincleavage loop as the receptor, and furin active pocket as the ligand. The protein-protein docking results showed that furin acidic/negative active pocket can be well fitted onto the SARS-CoV-2 Spikebasic/positive S1/S2 protease cleavage loop with low energy (-18.43 Kcal/mol). This implies that the extra “PRRAR” loop of SARS-CoV-2 Spike renders it more fragile to the protease. And this may allow this site to be cut during the maturation, efficiently
chinaXiv:202002.00062v1
enhancing the infection efficiency.
Figure 4.Protein-protein docking model of SARS-CoV-2 Spike with furin. (A) Superimposition of SARS-CoVSpike and SARS-CoV-2 Spike. Two S1/S2 protease cleavage sites and fusion peptide were shown as electrostatic surface mode. (B) Furin was docked onto the putative furin cut site (Arg685) of SARS-CoV-2 Spike. Both domains are shown as electrostatic surface mode.
3.3. Virtual ligand screening of furin protein
Structure-based virtual ligand screening method was used to screen potential furin protein inhibitors through ICM 3.7.3 modeling software (MolSoft LLC, San Diego, CA) from a ZINC Drug Database (2924 compounds), a small in-house database of natural products (including reported common antiviral components from traditional Chinese medicine) and derivatives (1066 compounds), and an antiviral compounds library contains 78 known antiviral drugs and reported antiviral compounds. Compounds with lower calculated binding energies (being expressed with scores and mfscores) are considered to have higher binding affinities with the target protein.
chinaXiv:202002.00062v1
The screening results for the ZINC Drug Database (Table 2) showed that anti-tumor drugs Aminopterin, Fludarabine phosphate and Irinotecan, antibacterial drugs Sulfoxone,Lomefloxacinand Cefoperazone,antifungaldrug Hydroxystilbamidine, antivirus drugValganciclovir,hepatoprotective drugSilybin,folic acid supplementFolinic acid have higher binding affinity to furin with mfscores lower than -100 or Scores lower than -30.
Here, we show one example of screen hits, Hydroxystilbamidine, which was predicted to bind in the active site of furin with low binding energy. In the generated docking model, Hydroxystilbamidine was well fitted into the binding pocket of the substrate and adopted similar conformation as substrate analogous inhibitor MI-52 in PDB model 5JXH[22],occupied two arms’ position of MI-52 (Figure 5A). Asp159, Asp259 and Asp306 were predicted to form three hydrogen bonds with imine groups of compounds (Figure 5B). It looks like that Hydroxystilbamidine mimic at least two arginines. Weak hydrophobic interaction between His194, Leu227, the backbone of Trp254 and Asn295 with the compound may further stabilize its conformation.
Table 2. Potential furin inhibitors from ZINC drug database
No.
Drug Name
Structure
Pharmacological functions
1
Aminopterin
Anti-tumor
2
Folic acid
Vitamin B9, necessary material for the growth and reproduction of body cells
3
Sulfoxone
Antibacterial effect
chinaXiv:202002.00062v1
4
Silybin
Fludarabine phosphate
Hepatoprotective effect
5
Diminazene
Insecticidal effect
6
Anti-tumor
7
L-Arginine
Nutritional supplement
8
Hydroxystilbamidine
Antifungal effect
9
Methotrexate
Antineoplastic, antirheumatic effects
10
L-dopa
Treatment of Parkinson’s disease
11
Irinotecan
Anti-tumor
chinaXiv:202002.00062v1
12
Cefoperazone
Valganciclovir
Antibacterial effect
13
Folinic acid
Folic acid supplement
14
Glycerol 3-phosphate
Intermediate for serine synthesis
15
Antivirus
16
Fosaprepitant
Treatment of nausea and vomiting induced by chemotherapy
17
Lomefloxacin
Antibacterial effect
18
Glutathione
Hepatoprotective effect
chinaXiv:202002.00062v1
19
Famotidine
Treatment of gastrohelcosis
20
Imatinib
Anti-tumor
21
Chenodeoxycholic acid
Dissolving gallstones
Figure 5.Low-energy binding conformations of Hydroxystilbamidine bound to furin generated by molecular docking. (A) Hydroxystilbamidinewas fitted well in the active pocket of human furin, and furin was shown as electrostatic surface model. Hydroxystilbamidine (yellow) was overlapped with substrate analogue inhibitor MI-52 (purple).(B) Detailed view of Hydroxystilbamidinebinding in the activepocket of furin.
chinaXiv:202002.00062v1
Another example was anticancer drug Imatinib. It was also predicted to bind in the active site of furin. In the generated docking model, Imatinib was fitted well in the binding pocket, and occupied the top two arms’ position of MI-52 (Figure 6A). Two hydrogen bonds were predicted to form between the compound with Glu236 and Gly255. Weak hydrophobic interaction between Val231, Pro256, Trp254 and Gly294 and the compound was found (Figure 6B).
Figure 6. Low-energy binding conformations of Imatinibto furin generated by molecular docking. (A) Imatinibwas fitted well in the active pocket of human furin, and furin was shown as electrostatic surface model. Imatinib (yellow) was overlapped with substrate analogue inhibitor MI-52 (purple).(B) Detailed view of Imatinibbinding in the activepocket of furin.
chinaXiv:202002.00062v1
Table 3.Potential furin inhibitors from in-house natural product database
No.
Drug Name
Structure
Pharmacological functions
Source
1
(-)-Epigallocatechin gallate
Antioxidation, anti-tumor, treatment of depression
For the natural products (Table 3), a series of compounds with antivirus and anti-inflammation effects, such as (-)-Epigallocatechin gallateand Theaflavin 3,3′-di-O-gallatefromCamellia sinensis,Biorobin from Ficusbenjamina, Andrographolide and 14-deoxy-11,12-didehydroandrographiside from Andrographispaniculata, one Andrographolide derivative (1S,2R,4aS,5R,8aS)-1- formamido-1,4a-dimethyl-6-methylene-5-((E)-2-(2-oxo-2,5-dihydrofuran-3-yl)ethenyl)decahydro
naphthalen-2-yl 5-((R)-1,2-dithiolan-3-yl)pentanoate, 3,4-seco-friedelolactone-27-lactone from Viola G7fromPhyllanthusemblica, xanthones2-[[2-O-(6-deoxy-α-L-mannopyranosyl)-β-D-xylopyranosyl]oxy]-1,8-dihydroxy-6-meth oxy-9H-xanthen-9-one, Kouitchenside J and Kouitchenside Ffrom Swertiakouitchensisexhibited high binding affinity to furin protein (mfscores< -100), suggesting the potential utility of these compounds in the treatment of SARS-CoV-2.
(-)-Epigallocatechin gallate (EGCG) was predicted to bind in the active site of furin, as Imatinib, it occupied the top two arms’ position of MI-52 (Figure 7A). Two hydrogen bonds were predicted formed between the compound with Asp258 and Ala292. Weak hydrophobic interactions between Pro256, Trp254 and Gly294 and the compound were predicted (Figure 7B).
diffusa,
2β,30β-dihydroxy- Phyllaemblicin three
chinaXiv:202002.00062v1
Figure 7. Low-energy binding conformations of ECCG to furin generated by molecular docking. (A) ECCGwas fitted well in the active pocket of human furin, and furin was shown as electrostatic surface model. ECCG (yellow) was overlapped with substrate analogue inhibitor MI-52 (purple).(B) Detailed view of ECCG binding in the activepocket of furin.
The database of 78 antiviral drugs including compounds already on the market and currently undergoing clinical trials to treat SARS-CoV-2 infections was further screened. The results were shown in Table 4. DNA topoisomerase II inhibitorSuramin treating hand-foot-and-mouth disease exhibited the highest affinity with furin (mfscore = 190.406). A series HIV-1 therapeutic drugs, such as Indinavir, Tenofoviralafenamide, TenofovirDisoproxil and Dolutegravir, and hepatitis C therapeutic drugs, Boceprevir and Telaprevir also have high binding affinity to furin.
Suramin was predicted to bind in the active site of furin with high binding mfScores. From generated docking model, Suramin occupied the top right arm and bottom arm positions of MI-52, it extended more to another adjacent pocket and covered almost all the surface areas for furin substrate binding (Figure 8A). Asp154, Asp228, Gly229, Ser253, Asp264, Glu271, Ile312, Lys449,
chinaXiv:202002.00062v1
Arg490 and Asp530 were predicted to form 10 hydrogen bonds with the compound. Weak hydrophobic interactions may form between His194, Leu227, Tyr308, Trp531 and A532 with the compound (Figure 8B).
Table 4.Potential furin inhibitors from the common antiviral drugs database
No.
Drug Name
Suramin
Structure
Pharmacological functions
1
DNA topoisomerase II inhibitor
2
Indinavir
Human immunodeficiency virus Protease (HIV PR)
3
Boceprevir
Hepatitis C virus Serine protease NS3/4A (HCV NS3/4A) Modulator
Hepatitis C virus Serine protease NS3/4A (HCV NS3/4A) Modulator
8
Dolutegravir
Human immunodeficiency virus Integrase (HIV IN)
9
Maraviroc
1.C-C chemokine receptor type 5 (CCR5) 2.CCR5 messenger RNA(CCR5 mRNA)
10
Cobicistat
Inhibitor of cytochrome P450 3A (CYP3A) enzymes
11
Stavudine triphosphate
Nucleoside analogue reverse transcriptase inhibitor used in the treatment of HIV infection
chinaXiv:202002.00062v1
Figure 8.Low-energy binding conformations of Suraminto furin generated by molecular docking. (A) Suraminwas fitted well in the active pocket of human furin, and furin was shown as electrostatic surface model. Suramin (yellow) was overlapped with substrate analogue inhibitor MI-52 (purple).(B) Detailed view of Suraminbinding in the activepocket of furin.
chinaXiv:202002.00062v1
4. Discussion
Our previous study(accepted by ActaPharmaceuticaSinica B) analyzed the amino acid composition of the RBD domain of the ACE2 receptor of SARS-CoV-2 and Bat-CoVRaTG13. We found that several key amino acids determining binding were mutated in SARS-CoV-2, which are more similar tothat of SARS-CoV.The calculation results show that in the same conformation as the SARS-CoV protein, the binding energy of SARS-CoV-2 and ACE2 receptors was a litter higher, but this result cannot fully explain the epidemiologically high contagion, so we speculate (1)the RBD domain of SARS-CoV-2 may have other conformations; (2) there may be other receptors; and (3) there are other mechanisms that enhance infectivity. During this manuscript was prepared, the Cryo-EM structure of SARS-CoV-2 Spike was solved[20]. Comparing thestructure of SARS-CoV-2 with the Spike structure of SARS-CoV, combined with biophysical detection, they found that SARS-CoV-2 binds more strongly to cellular ACE2 receptors. Furthermore, the just disclosed crystal structure of SARS-CoV-2 RBD-ACE2 complex showed a distinct conformational change in the key loop of complex binding interface. And the binding free energy calculation indicated a slightly stronger binding for SARS-CoV-2 RBD compared to that of SARS RBD. These results confirm our guess that the conformational change of the RBD domain of SARS-CoV-2 leads to stronger binding. However, stronger receptor binding still can’t fully explain the more infectious problem.
So we put forward these hypotheses: (1) SARS-CoV-2 can also bind to other receptors; (2) the lung may not be the earliest infection site; (3) SARS-CoV-2 is easier to cut and more easily fuse with cell membranes. Published in the Pubmed database, researchers performed RNA-seq analysis on tissue samples from 95 individuals’ 27 different tissues. The results showed that ACE2 protein was highly expressed in the small intestine and duodenum, but the expression level in lung tissue is low (Figure S5). However, we analyzed the expression of furin and found that it is distributed in various organs with little difference in expression level. Combined with the possible infection mechanism of SARS-CoV-2, the widespread distribution of furin increases the SARS-CoV-2 infection of other organs. The possibility of other organ attack is consistent with the multiple symptoms observed in clinic of COVID-19.
Based on these three conjectures, we compared the Spike sequences from SARS-CoV-2, SARS-CoV, MERS-CoV and Bat-CoVRaTG13, and found that anextra “PRRA”insert near the
chinaXiv:202002.00062v1
S1/S2 cleavage site. The “PRRA”insert and subsequent arginine (R) constitute a RRAR sequence that can be recognized and cleaved by furin-like proteases, which may be the reason why SARS-CoV-2 infection is stronger than SARS-CoV. What’s more, we performed a homologous alignment and phylogenetic analysis of the SARS-CoV-2 sequence, and found that “PRRA”insert did not appear at any other close relatives of SARS-CoV-2, indicating that this insertwas completely novel in this genus virus. The existence of such a motif may allow Spikes to be cut into S1 and S2 by furin-like proteases before maturity, but not separated, which provides S1 with the flexibility to change the conformation to better fit the host receptor. According to Simmons G et al. studies, overexpression of furin can increase the activity of SARS-CoVSpike, but it will not cause Spike to be cleaved [23].This is consistent with our prediction.
Furthermore, Glowacka Ietal.and Simmons Get al.studies have demonstrated that SARS-CoVSpikes can be activated by cleavage in two ways, including proteolytic activation by cathepsins B and L in host cells [24]. In addition, SARS-CoVSpike can be activated by TMPRSS2 cleavage on the host cell surface[6].What’s more, MERA-CoV, S1/S1 and S2’ cleavage sites cannot be cut by fruin[25].So we speculated that the activation of SARS-CoV-2 Spike can be through different protease cleavage pathways and these pathways can occur simultaneously in host cells. SARS-CoV-2 Spike can utilize host protease diversity to activate, which may explain the strong infectious capacity of SARS-CoV-2. As we can see in Figure 2, the Spike protein of SARS-CoV-2 can be cleaved at multiple stages, which greatly increases the efficiency of fusion. It is likely that the virus will fuse with the cell during endocytosis and release the genome. In addition, the binding ability of the cleaved Spike to the ACE2 receptor is also greatly enhanced [26].
According to our study, furin-like proteases may be potential drug targets for anti-SARS-CoV-2 treatment. At present, some peptide inhibitors have been developed and have good effects [27, 28]. To search potential inhibitors of furin-like proteases, we screened potential compounds from a ZINC drug database (2924 compounds), a small in-house database of natural products (1066 compounds), and existing antiviral drugs library (78 compounds) withfurinby virtual ligand screening. From the ZINC Drug Database, we found a series of anti-tumor, antibacterial, antivirus,hepatoprotective drugs, such as Aminopterin, Fludarabinephosphate,Sulfoxone,Irinotecan, Hydroxystilbamidine, Lomefloxacin, Cefoperazone, Valganciclovir,Imatinib, etc. might be used as furin inhibitors. For the natural products, some
chinaXiv:202002.00062v1
flavonoids, diterpenoids, and steroids with antivirus and anti-inflammation effects, such as ECCG, Biorobin, Phyllaemblicin G7, Andrographolide and its derivatives, and xanthonesfrom the Swertiagenus, etc.exhibited high binding affinity to furin protein. From the database of 78 antiviral drugs, a series of HIV-1, hepatitis C, and hand-foot-and-mouth disease therapeutic drugs, such as Indinavir, Tenofoviralafenamide, Tenofovir, Disoproxil, Dolutdegravir, Boceprevir, Telaprevir and Suraminalso showed high binding affinity to furin. These potentialfurin inhibitors and medicinal plants containing these compounds as major constituents might be useful for the treatment COVID-19. The further experiments to verify their efficiency in viro and in vivo will be carried out in our future studies. What’s more, combined administration of targeting different SARS-CoV-2 proteases with furin inhibitors may be an effective therapeutic strategy.
ACKNOWLEDGEMENTS
We acknowledge support from National Mega-project for Innovative Drugs (grant number 2019ZX09721001-004-007), National Natural Science Foundation of China (NSFC) (grant number No.U1803122, 81773637, 81773594, U1703111).
chinaXiv:202002.00062v1
5. References
[1] Bosch BJ, van der Zee R, de Haan CA, Rottier PJ. The coronavirus Spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J Virol 2003; 77: 8801-8811. [2] Lu G, Wang Q, Gao GF. Bat-to-human: Spike features determining ‘host jump’ of coronaviruses SARS-CoV, MERS-CoV, and beyond. Trends in Microbiology 2015; 23: 468-478.
[3] Xu X, Chen P, Wang J, Feng J, Zhou H, Li X, Zhong W, Hao P. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its Spike protein for risk of human transmission. Sci China Life Sci 2020.https://doi.org/10.1007/s11427-020-1637-5. [4] Letko M, Munster V. Functional assessment of cell entry and receptor usage for lineage B β-coronaviruses, including 2019-nCoV. bioRxiv, 2020.https://doi.org/10.1101/2020.01.22.915660.
[5] Belouzard S, Chu VC, Whittaker GR. Activation of the SARS coronavirus Spike protein via sequential proteolytic cleavage at two distinct sites. Proc Natl Acad Sci U S A 2009; 106: 5871-5876. [6] Glowacka I, Bertram S, Muller MA, Allen P, Soilleux E, Pfefferle S, Steffen I, Tsegaye TS, He Y, Gnirss K, Niemeyer D, Schneider H, Drosten C, Pohlmann S. Evidence that TMPRSS2 Activates the Severe Acute Respiratory Syndrome Coronavirus Spike Protein for Membrane Fusion and Reduces Viral Control by the Humoral Immune Response. Journal of Virology 2011; 85: 4122-4134.
[7] Zhou Y, Vedantham P, Lu K, Agudelo J, Carrion R, Jr., Nunneley JW, Barnard D, Pohlmann S, McKerrow JH, Renslo AR, Simmons G. Protease inhibitors targeting coronavirus and filovirus entry. Antiviral Res 2015; 116: 76-84. [8] Kam YW, Okumura Y, Kido H, Ng LFP, Bruzzone R. Ralf AltmeyerCleavage of the SARS Coronavirus Spike Glycoprotein by Airway Proteases Enhances Virus Entry into Human Bronchial Epithelial Cells In Vitro. PLoS ONE 2009; 4: e7870.
[9] Feliciangeli SF, Thomas L, Scott GK, Subbian E, Hung CH, Molloy SS, Jean F, Shinde U, Thomas G. Identification of a pH sensor in the furin propeptide that regulates enzyme activation. J Biol Chem 2006; 281: 16108-16116. [10] Henrich S, Cameron A, Bourenkov GP, Kiefersauer R, Huber R, Lindberg I, Bode W, Than ME. The Crystal Structure of the Proprotein Processing Proteinase Furin Explains Its Stringent Specificity. Nat Struct Biol 2003; 10: 520-526.
[11] Tay FP, Huang M, Wang L, Yamada Y, Liu DX. Characterization of cellular furin content as a potential factor determining the susceptibility of cultured human and animal cells to coronavirus infectious bronchitis virus
chinaXiv:202002.00062v1
infection. Virology 2012; 433: 421-430. [12] Yamada Y, Liu DX. Proteolytic activation of the Spike protein at a novel RRRR/S motif is implicated in furin-dependent entry, syncytium formation, and infectivity of coronavirus infectious bronchitis virus in cultured cells. J Virol 2009; 83: 8744-8758. [13] Matsuyama S, Shirato K, Kawase M, Terada Y, Kawachi K, Fukushi S, Kamitani W. Middle East Respiratory Syndrome Coronavirus Spike Protein Is Not Activated Directly by Cellular Furin during Viral Entry into Target Cells. J Virol 2018; 92. [14] Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, Coleman RG. ZINC: A Free Tool to Discover Chemistry for Biology. Journal of Chemical Information and Modeling 2012; 52: 1757-1768. [15] Abagyan R, Totrov M, Kuznetsov D. ICM-A New Method for Protein Modeling and Design_ Applications to Docking and Structure Prediction from the Distorted Native Conformation. J Comput Chem 1994; 15: 488-506 [16] Millet JK, Whittaker GR. Host cell proteases: Critical determinants of coronavirus tropism and pathogenesis. Virus Research 2015; 202: 120-134. [17] Walls AC, Xiong X, Park Y-J, Tortorici MA, Snijder J, Quispe J, Cameroni E, Gopal R, Dai M, Lanzavecchia A, Zambon M, Rey FA, Corti D, Veesler D. Unexpected Receptor Functional Mimicry Elucidates Activation of Coronavirus Fusion. Cell 2019; 176: 1026-1039. [18] Kirchdoerfer RN, Cottrell CA, Wang N, Pallesen J, Yassine HM, Turner HL, Corbett KS, Graham BS, McLellan JS, Ward AB. Pre-fusion structure of a human coronavirus Spike protein. Nature 2016; 531: 118-121. [19]http://nmdc.cn/?from=groupmessage#/resource/detail?no=NMDCS0000001 [20]Daniel Wrapp, Nianshuang Wang, Kizzmekia S. Corbett, Jory A. Goldsmith, Ching-Lin Hsieh, Olubukola Abiona, Barney S. Graham, View ORCID ProfileJason S. McLellan. Cryo-EM Structure of the 2019-nCoV Spike in the Prefusion Conformation. bioRxiv. 2020. https://doi.org/10.1101/2020.02.11.944462. [21] Yuan Y, Cao D, Zhang Y, Ma J, Qi J, Wang Q, Lu G, Wu Y, Yan J, Shi Y, Zhang X, Gao GF. Cryo-EM structures of MERS-CoV and SARS-CoV Spike glycoproteins reveal the dynamic receptor binding domains.Nat Commun. 2017; 10:15092. [22] Dahms SO, Arciniega M, Steinmetzer T, Huber R, Than ME. Structure of the unliganded form of the proprotein convertase furin suggests activation by a substrate-induced mechanism. Proc Natl Acad Sci 2016; 113:11196-11201. [23] Simmons G, Bertram S, Glowacka I, Steffen I, Chaipan C, Agudelo J, Lu K, Rennekamp AJ, Hofmann H, Bates P, Pohlmann S. Different host cell proteases activate the SARS-coronavirus Spike-protein for cell-cell and
chinaXiv:202002.00062v1
virus-cell fusion. Virology 2011; 413: 265-274. [24] Simmons G, Gosalia DN, Rennekamp AJ, Reeves JD, Diamond SL, Bates P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc Natl Acad Sci U S A 2005; 102: 11876-11881. [25] Van Lam van T, Ivanova T, Hardes K, Heindl MR, Morty RE, Bottcher-Friebertshauser E, Lindberg I, Than ME, Dahms SO, Steinmetzer T. Design, Synthesis, and Characterization of Macrocyclic Inhibitors of the Proprotein Convertase Furin. ChemMedChem 2019; 14: 673-685. [26] Parka J-E, Lib K, Barlana A, Fehrc AR, Perlmanb S, McCray PB Jr, Gallagher T. Proteolytic processing of Middle East respiratory syndrome coronavirus Spikes expands virus tropism.Proc Natl Acad Sci U S A 2016; 113: 12262–12267 [27] Dahms SO, Hardes K, Steinmetzer T, Than ME. X-ray Structures of the Proprotein Convertase Furin Bound with Substrate Analogue Inhibitors Reveal Substrate Specificity Determinants beyond the S4 Pocket. Biochemistry 2018; 57: 925-934. [28] Dahms SO, Jiao GS, Than ME. Structural Studies Revealed Active Site Distortions of Human Furin by a Small Molecule Inhibitor. ACS Chem Biol 2017; 12: 1211-1216.
chinaXiv:202002.00062v1
Furin, a potential therapeutic target for COVID-19 Supplementary information
Figure S1. Multiple sequence alignment of 1000 Spike proteins. These 156 proteins were ranked according to their homology with SARS-2.The sequence corresponding to PRRA in SARS-CoV-2 in each sequence is marked in the red box.
chinaXiv:202002.00062v1
Figure S2. Result of furin cleavage site pridiction of Spike protein in SARS-CoV-2, which predicted by online method ProP 1.0 Server.
Figure S3. Protein-protein dockingcalculation model of SARS-CoV-2 spike RBD (light blue) with human ACE2 (yellow), original RBD conformation was shown in orange. The calculated free energy is -50.13 Kcal/mol.
chinaXiv:202002.00062v1
Figure S4. Comparison of SARS-CoV-2 spike RBD (orange) and SARS spike RBD (yellow). The complex with ACE2 (left part, yellow) was shown. The homology model of SARS-CoV-2 spike RBDbuilt from SARS spike RBD was shown as blue.
chinaXiv:202002.00062v1
Figure S5. Expression levels of Furin, ACE2 and TMPRSS2 in various tissues. The data is from pubmed [1-3].
chinaXiv:202002.00062v1
References
[1] Angiotensin I converting enzyme 2 (ACE2) expression level in human tissues using HPA RNA-seq method. [DB/OL].(2020-02-03) [2020-02-17]. https://www.ncbi.nlm.nih.gov/gene/59272/?report=expression. [2] Furin, paired basic amino acid cleaving enzyme (Fruin) expression level in human tissues using HPA RNA-seq method. [DB/OL].(2019-12-21) [2020-02-17]. https://www.ncbi.nlm.nih.gov/gene/5045/?report=expression.
[3] Transmembrane serine protease 2 (TMPRSS2) expression level in human tissues using HPA RNA-seq method. [DB/OL].(2019-12-21) [2020-02-17]. https://www.ncbi.nlm.nih.gov/gene/7113?report=expression.
Potential treatment of Chinese and Western Medicine targeting nsp14 of 2019-nCoV
Chao Liu 1, Xiaoxiao Zhu 1, Yiyao Lu 1, Xu Jia 1*, Tai Yang 2*
1 Non-coding RNA and Drug Discovery Key Laboratory of Sichuan Province, Chengdu Medical College, Chengdu, Sichuan, China
2 School of Pharmacy, Chengdu Medical College, Chengdu, Sichuan, China *Correspondence to: Xu Jia: jiaxu@cmc.edu.cn
Tai Yang: taiyang@cmc.edu.cn
chinaXiv:202002.00071v2
Abstract
2019 novel coronavirus (2019-nCoV) caused severe, large-scale acute respiratory disease outbreak in Wuhan, China. The 2019-nCoV has spread to other regions and countries around the world, which is seriously threatening human health. There is an urgent need to develop drugs for the prevention and treatment of 2019-nCoV. 2019- nCoV nonstructural protein 14 (NSP14) carrying RNA cap guanine N7- methyltransferase and 3′-5′ exoribonuclease activities could be a potential drug target for intervention. NSP14 of 2019-nCoV shared 98.7% similarity with the one (PDB ID: 5nfy) of acute respiratory syndrome (SARS) Coronavirus. Then, the 2019-nCoV NSP14 structures were modelled by using SARS NSP14 (PDB ID: 5nfy) as template for virtual screening. Based on the docking score, 18 small molecule drugs were selected for further evaluation. The compounds, including Saquinavir, Hypericin, Baicalein and Bromocriptine, could bind the N-terminus and C-terminus of the homology model of the 2019-nCoV Nsp14, thus providing as a candidate drug against 2019-nCoV for further study.
chinaXiv:202002.00071v2
1. Introduction
In December 2019, a large scale, severe acute respiratory disease named as “2019 novel coronavirus (2019-nCoV)” occurred in Wuhan, China, and has already spread to other regions of China and other countries around the world in the following one month, seriously threatening human health. There is an urgent demand to develop drugs for the prevention and treatment of 2019-nCoV. Coronavirus NSP plays an important role in the virus’ genome replication and transcription 1,2, and it is generally conserved as an important functional protein in the coronavirus family. Among the family, NSP14 protein has both exonuclease and methyltransferase functions, which is important for replication and transcription of SARS and other coronavirus, thus providing attractive target for drug designs 3-5.
The amino acid sequence alignment revealed that the NSP14 of 2019-nCoV shared 98.7% similarity with the one (PDB ID: 5nfy) of SARS (Figure 1). Thus, the 2019- nCoV NSP14 structures were modelled by using SARS NSP14 (PDB ID: 5nfy) as template. The N-terminus and C-terminus of 2019-nCoV NSP14 were designated as active sites for screening drugs. A total of 7496 drugs obtained in the ZINC database were subjected to the binding screening. Among them, 2100 drugs were approved by FDA, 4264 drugs were approved by other regulatory agencies besides FDA (world-not- FDA) and 1132 drugs are undergoing clinical trials but not yet approved (investigational-only). The docking was carried out using AutoDock Vina1.1.2. Ten top compounds showed the lowest negative vina score in a range of -8.6 to -9.7 kcal/mol were selected from the N-terminal domain of homology model (Table. 1), and eight top compounds with lowest negative vina score in a range of -8.7 to -9.7 kcal/mol were achieved from the C-terminal domain of homology model (Table. 2). More importantly, the compounds, including Saquinavir, Hypericin, Baicalein and Bromocriptine, not only could bind the N-terminus and C-terminus of the homology model (Figure 1. A, B), but also could bind the N- terminal and C-terminal active pockets of the 2019-nCoV Nsp14 (Figure 1. C, D).
chinaXiv:202002.00071v2
2. Materials and Methods 2.1 Docking method
The SARS NSP14 amino acid sequence was downloaded from the PDB protein structure database (PDB ID: 5nfy). The 2019-nCoV amino acid sequence (Accession number: MN908947) was obtained from database of the National Center for Biotechnology Information (NCBI). The homology of above amino acid sequence was aligned using ClustalW. Homology model of the target protein was constructed and optimized by Modeller9.18 using crystal structure of SARS NSP14 (PDB ID: 5nfy) as template. A total of 100 independent structures were constructed, and the one with best DOPE score was selected for further energy minimization by Amber.
The ligands were downloaded from the ZINC database (FDA, world-not-FDA, investigational-only, http://zinc.docking.org/substances/subsets/). The 2D structure of the compound was then converted into the corresponding 3D coordinates using the Babel server (http://openbabel.sf.net). Then the model was converted to pdbqt format by prepare_receptor4.py script with assigning atom type and partial charge. All rotatable bonds in the ligand were set as flexible for flexible docking. Vina1.1.2 was used for molecular docking. The docking boxes were selected at the N-terminal exonuclease domain (aa: 62-290) and C-termimal transmethylase domain (aa: 291-527) of 2019-nCoV NSP14 respectively.
2.2 Binding free energy calculation
Each simulation system was immersed in a cubic box of TIP3P water with 10 Å distance from the solute. The Na+ or Cl- was applied to neutralize the system. General Amber force field (GAFF) 15 and Amber ff14SB force field were used to parameterize the ligand and protein respectively. 10,000 steps of minimization with constraints (10 kcal/mol/Å2) on heavy atoms of complex, including 5,000 steps of steepest descent minimization and 5,000 steps of conjugate gradient minimization, was used to optimize each system. Then each system was heated to 300 K within 0.2 ns followed by 0.1 ns equilibration in NPT ensemble. Finally, 5 ns MD simulation on each system at 300 K
chinaXiv:202002.00071v2
was performed. The minimization, heating and equilibrium are performed with sander program in Amber18. The 5 ns production run was performed with pmemd.cuda. Based on the 5 ns MD simulation trajectory, binding free energy (ΔG) was calculated with MM/GBSA method according to the following equation: ΔGcal=ΔH- TΔS=ΔEvdw+ΔEele+ΔGgb+ΔGnp-TΔS, where ΔEele and ΔEVDW refer to electrostatic and van der Waals energy terms respectively. ΔGgb and ΔGnp refer to polar and non-polar solvation free energies respectively. Conformational entropy (TΔS) was not calculated for saving time. Besides, the ligands were compared based on the same target, so it is reasonable to ignore the entropy.
3. Results and Discussion
3.1 Docking results of Saquinavir against 2019-nCoV NSP14 model
Saquinavir, as the first FDA-approved HIV protease inhibitor, has been used in the treatment of patients with human immunodeficiency virus (HIV) infection since 1995 6. Our docking results showed that five of the hydrogen bonds involving ASP-273, ASN-252, ASP-90, and LEU-253 were maintained upon the binding of Saquinavir and N terminus of 2019-nCoV NSP14 (Fig. 1A). Meanwhile hydrogen bonds involving ASN-386, GLN-313, GLY-333 and THR-428 maintained upon the binding of Saquinavir and C terminus of 2019-nCoV NSP14 (Fig. 1C). Saquinavir could bind to the N- and C-terminal active pockets of the 2019-nCoV NSP14 (Fig. 1B, D). The recent study from a drug-target interaction deep learning model showed that Saquinavir can bind to 2019-nCoV RNA-dependent RNA polymerase to inhibit the enzyme activity7. Our simulation results showed that Saquinavir can bind two active sites of NSP14, thus Saquinavir could be as a candidate drug against 2019-nCoV for further research.
3.2 Docking results of Hypericin against 2019-nCoV NSP14 model
Hypericin as a main ingredient in traditional Chinese medicine- Hypericum perforatum L. (St. John’s wort) has been demonstrated activity against RNA viruses in
chinaXiv:202002.00071v2
vitro by inhibiting viral replication 8. The present docking results showe that three of the hydrogen bonds involving ASN-252, GLY-93, and HIS-268 are maintained upon the binding of Hypericin and N-terminus of 2019-nCoV NSP14 (Fig. 2A). The six hydrogen bonds involving ASN-306, ARG-310, ASN-422 and LY-336 are maintained upon the binding of Hypericin and C-terminus of it (Fig. 2C). Hypericin can bind to the N- and C-terminal active pockets of the 2019-nCoV Nsp14 (Fig. 2B, D). Hypericin has been proven to have inhibitory effects on human hepatitis C virus (HCV) and human immunodeficiency virus (HIV)9. Combined the present study, Hypericin may have potential antiviral effect against 2019-NcoV. The traditional Chinese medicine- Hypericum perforatum L as main composition of Shuanghuanglian oral liquid has been widely used for the treatment of viral influenza. However, Shuanghuanglian oral solution has been suggested for the treatment of 2019-nCoV, triggering a huge crisis of public trust in Chinese scientists. We suggested that anti-2019-nCoV effects of Hypericin should be detected in cell culture models of 2019-nCoV infection. This will help us have a good understanding whether it is a good method to use Chinese medicines for the treatment of 2019-nCoV or not.
3.3 Docking results of Baicalein against 2019-nCoV NSP14 model
Baicalein, a flavonoid compound isolated from the root of Scutellaria baicalensis Georgi (Huang Qin in Chinese), inhibit viral replication of parainfluenza, influenza A, hepatitis B, HIV-1, and SARS coronavirus 10-12. The present docking results showed that six of the hydrogen bonds involving ASN-266, ASP-273, GLY-93, GLU-92, and HIS-268 were maintained upon the binding of Hypericin and N-terminus of 2019-nCoV NSP14 (Fig. 2A). Four hydrogen bonds involving ASN-386, ASP-331, and GLN-313 are maintained upon the binding of Hypericin and C-terminus of it (Fig. 2C). Baicalein can also bind to the N- and C-terminal active pockets of the 2019-nCoV NSP14 (Fig. 2B, D). The previous study showed that Baicalein as a novel chemical inhibitor could inhibit ATPase activity of NSP13 protein of SARS coronavirus 13. The present data suggests that Baicalin may bind to NSP14 protein to exert anti-2019-nCoV activity.
chinaXiv:202002.00071v2
Therefore, we suspect that the anti-2019-nCoV activity induced by the Baicalein could be valuable for further study.
3.4 Docking results of Bromocriptine against 2019-nCoV NSP14 model
Bromocriptine, a specific dopamine receptor agonist for the hypothalamus and pituitary, has inhibitory effect on replication the Dengue virus with low cytotoxicity (half maximal effective concentration, EC50=0.8-1.6 μM; and half maximal cytotoxicity concentration, CC50=53.6 μM) 14. Moreover, Bromocriptine inhibited Zika virus protease activities and exhibited synergistic effects with interferon-α2b against Zika virus replications15. It is interesting to find that Bromocriptine can bind to the N- and C-terminal active pockets of the 2019-nCoV NSP14 from our molecular docking results (Fig. 4B, D). The present results showed that three of the hydrogen bonds involving ASN-104, ASP-273 and GLN-145 were maintained upon the binding of hypericin and N terminus of 2019-nCoV NSP14 (Fig. 4A). There was one bond involving THR-428 maintained upon the binding of hypericin and C terminus of it (Fig. 4C).
3.5 The calculation of binding free energy
Based on the 5 ns MD simulation trajectory, binding free energy (ΔG) was calculated by MM/GBSA method. The calculated binding free energies of Saquinavir, Hypericin, Baicalein and Bromocriptine for the N-terminus of the homology model were -37.2711±3.2160, -30.1746±3.1914, -23.8953±4.4800, -34.1350±4.3683 kcal/mol, respectively (Table 3), while the calculated binding free energies were – 60.2757±4.7708, -30.9955±2.9975, -46.3099±3.5689, -59.8104±3.5389 respectively, when binding to the C-terminus (Table 4). Taken together, the results demonstrated that Saquinavir had the strong binding free energy.
4. Conclusion
2019-nCoV NSP14, a bifunctional enzyme carrying RNA cap guanine N7-
chinaXiv:202002.00071v2
methyltransferase and 3′-5′ exoribonuclease activities could be a potential drug target for intervention. 2019-nCoV NSP 14 shares 98.7% sequence similarity with the corresponding one in SARS. Thus, the homology models of 2019-nCoV NSP14 was structured for virtual screening. Based on the docking score, 18 drugs were selected for further evaluations. Four drugs (Saquinavir, Hypericin, Baicalein and Bromocriptine) could bind to the N-terminal and C-terminal domains of 2019-nCoV NSP 14. Combined the anti-viral function of above four drugs reported by the published literatures, we suggest the anti-2019-nCoV effects of above four drugs should be evaluated in the cell culture models of 2019-nCoV infection.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (NO. 31870135, 31600116) and the “1000 Talent Plan” of Sichuan Province (NO. 980).
References
1. Snijder EJ, Decroly E, Ziebuhr J. The Nonstructural Proteins Directing Coronavirus RNA Synthesis and Processing. Adv Virus Res 2016; 96: 59‐126.
2. Sawicki SG, Sawicki DL, Siddell SG. A contemporary view of coronavirus transcription. J Virol 2007; 81(1): 20‐9.
3. Chen Y, Tao J, Sun Y, et al. Structure‐function analysis of severe acute respiratory syndrome coronavirus RNA cap guanine‐N7‐methyltransferase. J Virol 2013; 87(11): 6296‐305.
4. Eckerle LD, Lu X, Sperry SM, Choi L, Denison MR. High fidelity of murine hepatitis virus replication is decreased in nsp14 exoribonuclease mutants. J Virol 2007; 81(22): 12135‐44.
5. Chen Y, Cai H, Pan J, et al. Functional screen reveals SARS coronavirus nonstructural protein nsp14 as a novel cap N7 methyltransferase. Proc Natl Acad Sci U S A 2009; 106(9): 3484‐9.
6. Kitchen VS, Skinner C, Ariyoshi K, et al. Safety and activity of saquinavir in HIV infection. Lancet 1995; 345(8955): 952‐5.
7. Beck BR, Shin B, Choi Y, Park S, Kang K. Predicting commercially available antiviral drugs that may act on the novel coronavirus (2019‐nCoV), Wuhan, China through a drug‐target interaction deep learning model. bioRxiv 2020.
8. Karioti A, Bilia AR. Hypericins as potential leads for new therapeutics. Int J Mol Sci 2010; 11(2): 562‐94.
9. Lenard J, Rabson A, Vanderoef R. Photodynamic inactivation of infectivity of human
chinaXiv:202002.00071v2
immunodeficiency virus and other enveloped viruses using hypericin and rose bengal: inhibition of fusion and syncytia formation. Proc Natl Acad Sci USA 1993; 90(1): 158‐62.
10. Zhang G‐H, Wang Q, Chen J‐J, Zhang X‐M, Tam S‐C, Zheng Y‐T. The anti‐HIV‐1 effect of scutellarin. Biochem Biophys Res Commun 2005; 334(3): 812‐6.
11. Sithisarn P, Michaelis M, Schubert‐Zsilavecz M, Cinatl J. Differential antiviral and anti‐inflammatory mechanisms of the flavonoids biochanin A and baicalein in H5N1 influenza A virus‐infected cells. Antiviral Res 2013; 97(1): 41‐8.
12. Hour MJ, Huang SH, Chang CY, et al. Baicalein, Ethyl Acetate, and Chloroform Extracts of Scutellaria baicalensis Inhibit the Neuraminidase Activity of Pandemic 2009 H1N1 and Seasonal Influenza A Viruses. Evid Based Complement Alternat Med 2013; 2013: 750803.
13. Keum Y‐S, Lee JM, Yu M‐S, Chin Y‐W, Jeong Y‐JJBotKCS. Inhibition of SARS Coronavirus Helicase by Baicalein. 2013; 34(11): 3187‐8.
14. Kato F, Ishida Y, Oishi S, et al. Novel antiviral activity of bromocriptine against dengue virus replication. Antiviral Res 2016; 131: 141‐7.
15. Chan JF, Chik KK, Yuan S, et al. Novel antiviral activity and mechanism of bromocriptine as a Zika virus NS2B‐NS3 protease inhibitor. Antiviral Res 2017; 141: 29‐37.
chinaXiv:202002.00071v2
Figure and Table legends
Figure 1. The binding model of Saquinavir against 2019-nCoV NSP14. (A) Interactions between Saquinavir (cyan) and associated residues (off-white) in the N-terminus of the homology model for 2019-nCoV; (B) Binding models of Saquinavir (cyan) in the 2019-nCoV NSP14 protein N-terminus pocket (white surface); (C) Interactions between Saquinavir (cyan) and associated residues (off- white) in the C-terminus of the homology model for 2019-nCoV; (D) Binding models of Saquinavir (cyan) in the 2019-nCoV NSP14 protein C-terminus pocket (white surface). Numbers accompanying dashed yellow lines represents the interaction distance (Å).
chinaXiv:202002.00071v2
Figure 2. The binding model of Hypericin against 2019-nCoV NSP14. (A) Interactions between Hypericin (cyan) and associated residues (off-white) in the N-terminus of the homology model for 2019-nCoV; (B) Binding models of Hypericin (cyan) in the 2019-nCoV NSP14 protein N-terminus pocket (white surface); (C) Interactions between Hypericin (cyan) and associated residues (off- white) in the C-terminus of the homology model for 2019-nCoV; (D) Binding models of Hypericin (cyan) in the 2019-nCoV NSP14 protein C-terminus pocket (white surface). Numbers accompanying dashed yellow lines represents the interaction distance (Å).
chinaXiv:202002.00071v2
Figure 3. The binding model of Baicalein against 2019-nCoV NSP14. (A) Interactions between Baicalein (cyan) and associated residues (off-white) in the N-terminus of the homology model for 2019-nCoV; (B) Binding models of Baicalein (cyan) in the 2019-nCoV NSP14 protein N-terminus pocket (white surface); (C) Interactions between Baicalein (cyan) and associated residues (off-white) in the C-terminus of the homology model for 2019-nCoV; (D) Binding models of Baicalein (cyan) in the 2019-nCoV NSP14 protein C-terminus pocket (white surface). Numbers accompanying dashed yellow lines represents the interaction distance (Å).
chinaXiv:202002.00071v2
s
Figure 4. The binding model of Bromocriptine against 2019-nCoV NSP14. (A) Interactions between Bromocriptine (cyan) and associated residues (off-white) in the N-terminus of the homology model for 2019-nCoV; (B) Binding models of Bromocriptine (cyan) in the 2019-nCoV NSP14 protein N- terminus pocket (white surface); (C) Interactions between Bromocriptine (cyan) and associated residues (off-white) in the C-terminus of the homology model for 2019-nCoV; (D) Binding models of Bromocriptine (cyan) in the 2019-nCoV NSP14 protein C-terminus pocket (white surface).
Numbers accompanying dashed yellow lines represents the interaction distance (Å).
chinaXiv:202002.00071v2
Table 1 –Ten drugs selected from the N-terminal domain of homology model
Table 1 –Ten drugs selected from the N-terminal domain of homology model
1 Department of Critical Care Medicine, Baoshan People’s Hospital, Baoshan 678000, China
2 Intervention and Cell Therapy Center, Peking University Shenzhen Hosptial, Shenzhen 518035, China
3 Yunnan Yasheng Medical Technology Co., Ltd., Kunming 650021, China
4 Emergency Department of the First Affiliated Hospital of Kunming Medical University, EICU/MICU, Kunming 650032, China
5 Yunnan Key Laboratory for Basic Research on Bone and Joint Diseases & Yunnan Stem Cell Translational Research Center, Kunming University, Kunming 650214, China
6 Department of Neonatology, Baoshan People’s Hospital, Baoshan, 678000, China
7 School of Pharmaceutical Sciences (Shenzhen), Sun Yat-sen University, Shenzhen 517108, China
8 Yunnan Jici Institute for Regenerative Medicine co., Ltd., Kunming 650106, China
# These authors contribute equally.
* Correspondence to: E-mail: huminynkm@163.com (Hu M.); qianchuanyun@126.com (Qian
C.); yfgao@hotmail.com (Gao Y.)
1
chinaXiv:202002.00084v1
Abstract
Background
The COVID-19 cases increased very fast in the last two months. The mortality among critically ill patients, especially the elder ones, was relatively high. Considering that most of the dead patients were caused by severe inflammation response, it is very urgent to develop effective therapeutic agents and strategies for these patients. The human umbilical cord mesenchymal stem cells (hUCMSCs) have shown very good capability to modulate immune response and repair the injured tissue with good safety.
Case Presentation
Here, we reported the treatment process and clinical outcome of a 65-year-old female critically ill COVID-19 patient infected with 2019-nCoV (now called SARS-CoV-2). The significant clinical outcome and well tolerance was observed by the adoptive transfer of allogenic hUCMSCs.
Conclusions
Our results suggested that the adoptive transfer therapy of hUCMSCs might be an ideal choice to be used or combined with other immune modulating agents to treat the critically ill COVID- 19 patients.
In December 2019, the outbreak of 2019 novel coronavirus (now called SARS-CoV-2) infected pneumonia (COVID-19) began in Wuhan, China. As of 22th Feb. 2020, 2019 novel coronavirus had infected 76392 people in China (among which 2348 were killed) and 1404 people in other twenty-seven countries and regions (among which twelve people were killed) [1]. It was reported that the elder patients were inclined to get more severe symptom, and the ICU admission ratio of them was significantly than the younger. Including the ground glass opacity in the lung, the other typical diagnosis characteristic of the critically ill patients was significant decrease in lymphocytes along with the increase of neutrophils. The ICU admission patients have higher concentrations of IL-6, G-CSF, IP10, MCP-1, MIP1A, and TNF-α, indication the occurrence of cytokine storm [2, 3]. Persistence of cytokine storm will thus cause the severe organ injury and death [4]. There are no good choice to overcome the cytokine storm, these critically ill patients were always treated with glucocorticoid. But in most cases, the treatment of glucocorticoid will cause severe side effects including osteoporosis and hypoimmunity, or even delay the clearance of the virus [2, 5]. Therefore, it is very urgent to discover novel strategies to treat these critically ill patients [6].
Mesenchymal stem cells (MSCs) have been widely used to treat type 2 diabetes, autoimmune disease, spinal cord injury, GVHD, and other diseases with very good safety [7, 8]. Among which, the umbilical cord mesenchymal stem cells (hUCMSCs) can be easily get and cultured. hUCMSCs have shown very significant immunomodulation and tissue repair effects with low immunogenicity, which makes them very ideal candidate to the allogenic adoptive transfer therapy. It was also suggested to be potential to treat the H5N1 infection induced acute lung injury, which showed similar inflammatory cytokine profile to that of COVID-19 [9]. Up to now, the therapeutic effects of MSCs on COVID-19 have not been reported yet.
Here, we will introduce a critically ill elder female patient in China infected with 2019 novel coronavirus. The characteristics of the vital signs, CT images, clinical laboratory profiles, and major immune cell changes will be investigated. The clinical outcome of hUCMSCs adoptive transfer therapy will be also discussed.
Case Presentation
3
chinaXiv:202002.00084v1
On January 27, 2020, a 65-year-old woman felt fatigue and fever with a body temperature of 38.2oC, then cough with small amount of white bubble sputum. Considering that she had flown from Wuhan on January 21, 2020, she was immediately sent to the Longling People’s Hospital, and the throat swabs were collected. Then, antibiotics and phlegm reducing drugs were given for supportive treatment. On January 28, she had chest tightness with SPO2 of 81%, and blood pressure 160/91 mmHg. On the same day, the real-time RT-PCR result reported 2019 novel coronavirus positive, and X-ray examination showed ground glass opacity in the right lung. IFN-α inhalation treatment was performed. On January 29 morning, she felt chest tightness and more difficult to breathe, along with shortness of breath. In the afternoon, she was admitted to the infectious disease department of the Baoshan People’s Hospital (a tertiary hospital near Longling County) for better treatment.
On January 29, the clinical laboratory examination showed that the white blood cell count was in normal range, but the neutrophil percentage was increased to 87.9%, along with the lymphocyte percentage decreased to 9.8%. According to the guideline for the diagnosis and treatment of 2019 novel coronavirus infected pneumonia (Trial 4th Edition), the patient was treated with antiviral therapy of lopinavir/ritonavir, IFN-α inhalation and oseltamivir (oseltamivir was withdrawn after once administration), and also intravenous injection of moxifloxacin, Xuebijing, methylprednisolone, and immunoglobulin. To reduce hypoxia and prevent respiratory muscle fatigue of the patient, the non-invasive mechanical ventilator was used under the advice and guidance of hospital specialist group.
On January 30, the patient could breathe easily under the ventilator, along with normal body temperature but paroxysmal cough. Considering that she got a severe diarrhea from January 30 night to January 31 morning, electrolyte replacement and rehydration were given for supportive treatment. In case of reducing the blood glucose level (postprandial glucose level around 9.6- 14.6 mM), insulin was given intramuscularly. On January 31, the diarrhea symptom reduced significantly, but the patient showed severe electrolyte disturbance. The white blood cell count increased to 12.16×109/L, among which the neutrophil percentage increased to 92.4%. The C- reaction protein increased to 44.64mg/L, along with erythrocyte sedimentation rate increased to 88mm/h. Under the cooperation of multi-discipline team, the patient was diagnosed as
critically ill type COVID-19 along with acute respiratory failure and acute diarrhea. Diabetes
4
chinaXiv:202002.00084v1
and hypertension remained to be further determined.
On February 1, the patient showed no diarrhea and no shortness of breath when stay calmly,
but with paroxysmal cough and small amount of white sputum. The white blood cell count continuously increased to 13.92×109/L, among which the neutrophil percentage increased to 95.1% and lymphocyte decreased to 2.9%. From February 1 evening to February 2 morning, the patient began to breathe fast with a respiratory rate of 35-44/min, which could not be improved by adjusting the parameter of the ventilator. The blood oxygen saturation was continuously lower than 86‒90%. Under the guidance of the COVID-19 specialist team, the patient was urgently transferred to the ICU, and invasive tracheal cannula was performed to decrease the respiratory distress.
From February 2 to 4, the white blood cell count slightly decreased, among which the percentage of neutrophil increased to 82.2% and lymphocyte to 12.5% (both were still abnormal). In the early morning of February 4, the patient got a gastrorrhagia with a liquid amount around 230mL. Considering the low levels of red blood cell count and hemoglobin, anemia symptom was shown which might be caused by immune or inflammation related hemolysis. To modulate immune cell ratio, thymosin α1 was given from February 3. Although a blood transfusion was performed on February 4, the red blood cell count (2.76×1012/L) and hemoglobin concentration (92.00g/L) were still very low on February 5. On February 6, the serum bilirubin continuously increased, with the concentrations of DBil to 43.8μM and I-Bil to 29.5μM, indicating liver injury possibility. The concentrations of CRP (82.69mg/L), PCT (0.102ng/mL), D-Dimer (4.76 μg/mL), and ProBNP (670.2pg/mL) were very high. Although the white cell count was in normal range (8.38×109/L), but the neutrophil percentage began to increase again to a very high level (92.4%). All these results indicated that the anti- inflammatory effects of glucocorticoid, antiviral drugs and antibiotics might not work very well, and the gastrorrhagia was suspected to be caused by the side effects of glucocorticoid.
On February 7, considering the severe organ injury caused by inflammatory response and side effects, the glucocorticoid and antiviral therapy were withdrawn under the advice and guidance of the specialist group. The hUCMSCs adoptive transfer therapy was proposed. On February 8, the physical condition of the patient was re-evaluated. It was confirmed that she
was critically ill type COVID-19, with severe pneumonia (mixed type), acute respiratory
5
chinaXiv:202002.00084v1
distress, multi-organ injury (liver, respiratory system, and blood), moderate anemia, hypertension, type 2 diabetes, electrolyte disturbance, immunosuppression, acute gastrointestinal bleeding, and other symptoms. The family member and patient agreed to try hUCMSCs adoptive transfer therapy. The therapeutic scheme was then discussed and approved by the ethics committee of the hospital and consent forms were signed by the family member before the therapy.
Discussion
As shown in Figure 1, the allogenic hUCMSCs produced under GMP condition were administrated intravenously for three times (5×107cells each time) on February 9, 12, and 15. During the therapy, antibiotics were given to prevent infection, and thymosin α1 was also given. After the first time adoptive transfer, no obvious side effects were observed, indicating it was well tolerated. As shown in Table 1, after the second administration, the serum bilirubin, CRP, and ALT/AST were gradually reduced, along with some other vital signs were also improved. The trachea cannula was also pulled off and the patient could ambulate on the ground from February 13 as well. As shown in Figure 2, after the second administration, the white blood cell count and neutrophil count decreased to the normal level, along with the lymphocyte count increased to normal level as well. More importantly, the counts of CD3+ T cell, CD4+ T cell, and CD8+ T cell were also remarkably increased to normal levels. It was also suggested that the immune modulating effects of thymosin α1 alone (From day 7 to day 12 in Figure 2) might be not very significant, indicating that hUCMSCs or combined with thymosin α1 could greatly reduce the inflammation response and help the recovery of antiviral immune cells and organs. Considering the characteristics of hUCMSCs, we speculated that they might homing to repair the injured tissues and neutralize the inflammatory cytokines (such as G-CSF and IL-6) by the expression of their receptors.
As shown in Figure 3, by comparing the chest CT images taken on January 29 to February 16 and February 21, it can be seen that the pneumonia was greatly relieved. On February 17, the patient was transferred out of ICU, and most of the vital signs and clinical laboratory indexes recovered to normal level. The throat swabs tests reported negative on both February 17 and February 19.
6
chinaXiv:202002.00084v1
Conclusions
As a conclusion, we proposed that the adoptive transfer therapy of hUCMSCs might be an ideal choice to be used or combined with other immune modulating agents. Although only one case was shown here, it would also be very important to inspire more similar clinical practice to treat such critically ill COVID-19 patients.
Ethical Approval and Consent to participate
The study was approved by the Ethics Committee of the Baoshan People’s Hospital and informed consent was confirmed by the participants.
Consent for publication
Informed consent for publication was obtained from all authors and participants.
Availability of supporting data
The data supporting the results were included within the article.
Competing interests
All authors declare no competing interests.
Funding
This study was supported by the grants from Shenzhen Municipal Health Commission (SZSM201612071), the Ministry of Science and Technology of China (YCZYPT[2018] 03-1), and the Yunnan Science & Technology (2016RA093, 2018ZF007-03, 2019ZF002).
Authors’ contributions
Liang B., Li T., and Hu M. had full access to all of the data in the study and take responsibility for the accuracy of the data. Chen J. Wu H. and Qian C. analyzed the data. Gao Y. wrote the manuscript. Yang W., Li Y., Li J., Nie F., Ma Z., Yang M., and Nie P. participated the study and help to collect the data.
7
chinaXiv:202002.00084v1
Acknowledgements
We thank the grants supported by the Shenzhen Municipal Health Commission (SZSM201612071), the Ministry of Science and Technology of China (YCZYPT[2018] 03-1), and the Yunnan Science & Technology (2016RA093, 2018ZF007-03, 2019ZF002).
Authors’ information
Bing Liang#, Wenjie Yang#, Jianchun Li, Fangang Nie, Mingxi Yang
Department of Critical Care Medicine, Baoshan People’s Hospital, Baoshan 678000, China Junhui Chen#, Congtao Yu
Intervention and Cell Therapy Center, Peking University Shenzhen Hosptial, Shenzhen 518035, China
Tao Li#
Yunnan Yasheng Medical Technology Co., Ltd., Kunming 650021, China
Haiying Wu#, Chuanyun Qian*
Emergency Department of the First Affiliated Hospital of Kunming Medical University, EICU/MICU, Kunming 650032, China
Yanjiao Li, Zhaoxia Ma
Yunnan Key Laboratory for Basic Research on Bone and Joint Diseases & Yunnan Stem Cell Translational Research Center, Kunming University, Kunming 650214, China
Panrong Nie
Department of Neonatology, Baoshan People’s Hospital, Baoshan, 678000, China
Yanfeng Gao*
School of Pharmaceutical Sciences (Shenzhen), Sun Yat-sen University, Shenzhen 517108, China; Intervention and Cell Therapy Center, Peking University Shenzhen Hosptial, Shenzhen 518035, China
Min Hu*
Yunnan Key Laboratory for Basic Research on Bone and Joint Diseases & Yunnan Stem Cell Translational Research Center, Kunming University, Kunming 650214, China; Intervention and
Cell Therapy Center, Peking University Shenzhen Hosptial, Shenzhen 518035, China; Yunnan
8
chinaXiv:202002.00084v1
Jici Institute for Regenerative Medicine co., Ltd., Kunming 650106, China
# These authors contribute equally.
* Correspondence to: E-mail: huminynkm@163.com (Hu M.); qianchuanyun@126.com (Qian
C.); yfgao@hotmail.com (Gao Y.)
References
Data were cited from https://news.qq.com/zt2020/page/feiyan.htm.
Y, Wu W, Xie X, Yin W, Li H, Liu M, Xiao Y, Gao H, Guo L, Xie J, Wang G, Jiang R, Gao Z, Jin Q, Wang J, Cao B. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020; doi: 10.1016/S0140-6736(20)30183-5.
WangD,HuB,HuC,ZhuF,LiuX,ZhangJ,WangB,XiangH,ChengZ,XiongY,ZhaoY,LiY,Wang X, Peng Z. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA. 2020; doi: 10.1001/jama.2020.1585.
XuZ,ShiL,WangY,ZhangJ,HuangL,ZhangC,LiuS,ZhaoP,LiuH,ZhuL,TaiY,BaiC,GaoT, Song J, Xia P, Dong J, Zhao J, Wang F. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med. 2020; Pii: S2213-2600(20)30076-X.
Shang L, Zhao J, Hu Y, Du R, Cao B. On the use of corticosteroids for 2019-nCoV pneumonia. Lancet. 2020; pii: S0140-6736(20)30361-5.
Zumla A, Hui DS, Azhar EI, Memish ZA, Maeurer M. Reducing mortality from 2019-nCoV: host- directed therapies should be an option. Lancet. 2020; doi: 10.1016/S0140-6736(20)30305-6.
Le Blanc K, Rasmusson I, Sundberg B, Götherström C, Hassan M, Uzunel M, Ringdén O. Treatment of severe acute graft-versus-host disease with third party haploidentical mesenchymal stem cells. Lancet. 2004; 363(9419):1439-41.
Fan XL, Zhang Y, Li X, Fu QL. Mechanisms underlying the protective effects of mesenchymal stem cell-based therapy. Cell Mol Life Sci. 2020; doi:10.1007/s00018-020-03454-6.
Darwish I, Mubareka S, Liles WC. Immunomodulatory therapy for severe influenza. Expert Rev Anti Infect Ther. 2011; 9(7):807-22.
9
chinaXiv:202002.00084v1
Figure 1. The major symptoms and treatment of the critically ill COVID-19 patient
chinaXiv:202002.00084v1
Table 1. The major clinical laboratory characteristics of the patient
Days after disease onset
range 3 6 8 10 12 13* 14 16* 17 19* 21
Figure 2. The dynamic changes of the immune cell counts of the patient. The arrows indicate the day of hUCMSCs therapy. To the white blood cell (normal range 3.5‒9.5×109/L) and neutrophil (normal range 1.8‒6.3×109/L), the dash line indicates upper threshold. While to the lymphocyte (normal range 1.1‒3.2×109/L) and T cell subsets, the dash line indicates lower threshold.
chinaXiv:202002.00084v1
Figure 3. The typical CT images of the lung. A1-A3) CT images on January 29 indicate that there are lesions and mass density increasing shadow in both left and right lung. The ground- glass opacity, nonhomogeneous density, and air bronchus can be seen in the right lung. B1-B3) CT images on February 16 indicate the significant relief in both left and right lung. Only some Stripe shadow and small pieces of ground-glass opacity can be seen. C1-C3) CT images on February 21 indicate the further relief in both left and right lung. Most of the ground-glass opacity lightened, or even disappeared.
bioRxiv preprint first posted online Jan. 31, 2020; doi: http://dx.doi.org/10.1101/2020.01.30.927871. The copyright holder for this preprint (which was not peer-reviewed) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC-ND 4.0 International license.
Uncanny similarity of unique inserts in the 2019-nCoV spike protein to HIV-1 gp120 and Gag
1Kusuma School of biological sciences, Indian institute of technology, New Delhi-110016, India. 2Acharya Narendra Dev College, University of Delhi, New Delhi-110019, India
$Equal contribution
We are currently witnessing a major epidemic caused by the 2019 novel coronavirus (2019- nCoV). The evolution of 2019-nCoV remains elusive. We found 4 insertions in the spike glycoprotein (S) which are unique to the 2019-nCoV and are not present in other coronaviruses. Importantly, amino acid residues in all the 4 inserts have identity or similarity to those in the HIV- 1 gp120 or HIV-1 Gag. Interestingly, despite the inserts being discontinuous on the primary amino acid sequence, 3D-modelling of the 2019-nCoV suggests that they converge to constitute the receptor binding site. The finding of 4 unique inserts in the 2019-nCoV, all of which have identity /similarity to amino acid residues in key structural proteins of HIV-1 is unlikely to be fortuitous in nature. This work provides yet unknown insights on 2019-nCoV and sheds light on the evolution and pathogenicity of this virus with important implications for diagnosis of this virus.
Introduction
Coronaviruses (CoV) are single-stranded positive-sense RNA viruses that infect animals and humans. These are classified into 4 genera based on their host specificity: Alphacoronavirus, Betacoronavirus, Deltacoronavirus and Gammacoronavirus (Snijder et al., 2006). There are seven known types of CoVs that includes 229E and NL63 (Genus Alphacoronavirus), OC43, HKU1, MERS and SARS (Genus Betacoronavirus). While 229E, NL63, OC43, and HKU1 commonly infect humans, the SARS and MERS outbreak in 2002 and 2012 respectively occurred when the virus crossed-over from animals to humans causing significant mortality (J. Chan et al., n.d.; J. F. W. Chan et al., 2015). In December 2019, another outbreak of coronavirus was reported from Wuhan, China that also transmitted from animals to humans. This new virus has been temporarily termed as 2019-novel Coronavirus (2019-nCoV) by the World Health Organization (WHO) (J. F.- W. Chan et al., 2020; Zhu et al., 2020). While there are several hypotheses about the origin of 2019-nCoV, the source of this ongoing outbreak remains elusive.
The transmission patterns of 2019-nCoV is similar to patterns of transmission documented in the previous outbreaks including by bodily or aerosol contact with persons infected with the virus.
bioRxiv preprint first posted online Jan. 31, 2020; doi: http://dx.doi.org/10.1101/2020.01.30.927871. The copyright holder for this preprint (which was not peer-reviewed) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC-ND 4.0 International license.
Casesofmildtosevereillness,anddeathfromtheinfectionhavebeenreportedfromWuhan. This outbreak has spread rapidly distant nations including France, Australia and USA among others. The number of cases within and outside China are increasing steeply. Our current understanding is limited to the virus genome sequences and modest epidemiological and clinical data. Comprehensive analysis of the available 2019- nCoV sequences may provide important clues that may help advance our current understanding to manage the ongoing outbreak.
The spike glycoprotein (S) of cornonavirus is cleaved into two subunits (S1 and S2). The S1 subunit helps in receptor binding and the S2 subunit facilitates membrane fusion (Bosch et al., 2003; Li, 2016). The spike glycoproteins of coronoviruses are important determinants of tissue tropism and host range. In addition the spike glycoproteins are critical targets for vaccine development (Du et al., 2013). For this reason, the spike proteins represent the most extensively studied among coronaviruses. We therefore sought to investigate the spike glycoprotein of the 2019-nCoV to understand its evolution, novel features sequence and structural features using computational tools.
Methodology
Retrieval and alignment of nucleic acid and protein sequences
We retrieved all the available coronavirus sequences (n=55) from NCBI viral genome database (https://www.ncbi.nlm.nih.gov/) and we used the GISAID (Elbe & Buckland-Merrett, 2017)[https://www.gisaid.org/] to retrieve all available full-length sequences (n=28) of 2019- nCoV as on 27 Jan 2020. Multiple sequence alignment of all coronavirus genomes was performed by using MUSCLE software (Edgar, 2004) based on neighbour joining method. Out of 55 coronavirus genome 32 representative genomes of all category were used for phylogenetic tree development using MEGAX software (Kumar et al., 2018). The closest relative was found to be SARS CoV. The glycoprotein region of SARS CoV and 2019-nCoV were aligned and visualized using Multalin software (Corpet, 1988). The identified amino acid and nucleotide sequence were aligned with whole viral genome database using BLASTp and BLASTn. The conservation of the nucleotide and amino acid motifs in 28 clinical variants of 2019-nCoV genome were presented by performing multiple sequence alignment using MEGAX software. The three dimensional structure of 2019-nCoV glycoprotein was generated by using SWISS-MODEL online server (Biasini et al., 2014) and the structure was marked and visualized by using PyMol (DeLano, 2002).
Results
Uncanny similarity of novel inserts in the 2019-nCoV spike protein to HIV-1 gp120 and Gag
Our phylogentic tree of full-length coronaviruses suggests that 2019-nCoV is closely related to SARS CoV [Fig1]. In addition, other recent studies have linked the 2019-nCoV to SARS CoV. We therefore compared the spike glycoprotein sequences of the 2019-nCoV to that of the SARS CoV (NCBI Accession number: AY390556.1). On careful examination of the sequence alignment we found that the 2019- nCoV spike glycoprotein contains 4 insertions [Fig.2]. To further investigate if these inserts are present in any other corona virus, we performed a multiple
bioRxiv preprint first posted online Jan. 31, 2020; doi: http://dx.doi.org/10.1101/2020.01.30.927871. The copyright holder for this preprint (which was not peer-reviewed) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC-ND 4.0 International license.
sequence alignment of the spike glycoprotein amino acid sequences of all available coronaviruses (n=55) [refer Table S.File1] in NCBI refseq (ncbi.nlm.nih.gov) this includes one sequence of 2019-nCoV[Fig.S1]. We found that these 4 insertions [inserts 1, 2, 3 and 4] are unique to 2019-nCoV and are not present in other coronaviruses analyzed. Another group from China had documented three insertions comparing fewer spike glycoprotein sequences of coronaviruses . Another group from China had documented three insertions comparing fewer spike glycoprotein sequences of coronaviruses (Zhou et al., 2020).
Figure 1: Maximum likelihood genealogy show the evolution of 2019- nCoV: The evolutionary history was inferred by using the Maximum Likelihood method and JTT matrix-based model. The tree with the highest log likelihood (12458.88) is shown. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using a JTT model, and then selecting the topology with superior log likelihoodbioRxiv preprint first posted online Jan. 31, 2020; doi: http://dx.doi.org/10.1101/2020.01.30.927871. The copyright holder for this preprint (which was not peer-reviewed) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC-ND 4.0 International license.
value. This analysis involved 5 amino acid sequences. There were a total of 1387 positions in the final dataset. Evolutionary analyses were conducted in MEGA X.
Figure 2: Multiple sequence alignment between spike proteins of 2019-nCoV and SARS. The sequences of spike proteins of 2019-nCoV (Wuhan-HU-1, Accession NC_045512) and of SARS CoV (GZ02, Accession AY390556) were aligned using MultiAlin software. The sites of difference are highlighted in boxes.
We then analyzed all available full-length sequences (n=28) of 2019-nCoV in GISAID (Elbe & Buckland-Merrett, 2017) as on January 27, 2020 for the presence of these inserts. As most of these sequences are not annotated, we compared the nucleotide sequences of the spike glycoprotein of all available 2019-nCoV sequences using BLASTp. Interestingly, all the 4 insertions were absolutely (100%) conserved in all the available 2019- nCoV sequences analyzed [Fig.S2, Fig.S3].
bioRxiv preprint first posted online Jan. 31, 2020; doi: http://dx.doi.org/10.1101/2020.01.30.927871. The copyright holder for this preprint (which was not peer-reviewed) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC-ND 4.0 International license.
We then translated the aligned genome and found that these inserts are present in all Wuhan 2019- nCoV viruses except the 2019-nCoV virus of Bat as a host [Fig.S4]. Intrigued by the 4 highly conserved inserts unique to 2019-nCoV we wanted to understand their origin. For this purpose, we used the 2019-nCoV local alignment with each insert as query against all virus genomes and considered hits with 100% sequence coverage. Surprisingly, each of the four inserts aligned with short segments of the Human immunodeficiency Virus-1 (HIV-1) proteins. The amino acid positions of the inserts in 2019-nCoV and the corresponding residues in HIV-1 gp120 and HIV-1 Gag are shown in Table 1. The first 3 inserts (insert 1,2 and 3) aligned to short segments of amino acid residues in HIV-1 gp120. The insert 4 aligned to HIV-1 Gag. The insert 1 (6 amino acid residues) and insert 2 (6 amino acid residues) in the spike glycoprotein of 2019-nCoV are 100% identical to the residues mapped to HIV-1 gp120. The insert 3 (12 amino acid residues) in 2019- nCoV maps to HIV-1 gp120 with gaps [see Table 1]. The insert 4 (8 amino acid residues) maps to HIV-1 Gag with gaps.
Although, the 4 inserts represent discontiguous short stretches of amino acids in spike glycoprotein of 2019-nCoV, the fact that all three of them share amino acid identity or similarity with HIV-1 gp120 and HIV-1 Gag (among all annotated virus proteins) suggests that this is not a random fortuitous finding. In other words, one may sporadically expect a fortuitous match for a stretch of 6-12 contiguous amino acid residues in an unrelated protein. However, it is unlikely that all 4 inserts in the 2019-nCoV spike glycoprotein fortuitously match with 2 key structural proteins of an unrelated virus (HIV-1).
The amino acid residues of inserts 1, 2 and 3 of 2019-nCoV spike glycoprotein that mapped to HIV-1 were a part of the V4, V5 and V1 domains respectively in gp120 [Table 1]. Since the 2019- nCoV inserts mapped to variable regions of HIV-1, they were not ubiquitous in HIV-1 gp120, but were limited to selected sequences of HIV-1 [ refer S.File1] primarily from Asia and Africa.
The HIV-1 Gag protein enables interaction of virus with negatively charged host surface (Murakami, 2008) and a high positive charge on the Gag protein is a key feature for the host-virus interaction. On analyzing the pI values for each of the 4 inserts in 2019-nCoV and the corresponding stretches of amino acid residues from HIV-1 proteins we found that a) the pI values were very similar for each pair analyzed b) most of these pI values were 10±2 [Refer Table 1] . Of note, despite the gaps in inserts 3 and 4 the pI values were comparable. This uniformity in the pI values for all the 4 inserts merits further investigation.
As none of these 4 inserts are present in any other coronavirus, the genomic region encoding these inserts represent ideal candidates for designing primers that can distinguish 2019-nCoV from other coronaviruses.
bioRxiv preprint first posted online Jan. 31, 2020; doi: http://dx.doi.org/10.1101/2020.01.30.927871. The copyright holder for this preprint (which was not peer-reviewed) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC-ND 4.0 International license.
Table 1: Aligned sequences of 2019-nCoV and gp120 protein of HIV-1 with their positions in primary sequence of protein. All the inserts have a high density of positively charged residues. The deleted fragments in insert 3 and 4 increase the positive charge to surface area ratio. *please see Supp. Table 1 for accession numbers
The novel inserts are part of the receptor binding site of 2019-nCoV
To get structural insights and to understand the role of these insertions in 2019-nCoV glycoprotein, we modelled its structure based on available structure of SARS spike glycoprotein (PDB: 6ACD.1.A). The comparison of the modelled structure reveals that although inserts 1,2 and 3 are at non-contiguous locations in the protein primary sequence, they fold to constitute the part of glycoprotein binding site that recognizes the host receptor (Kirchdoerfer et al., 2016) (Figure 4). The insert 1 corresponds to the NTD (N-terminal domain) and the inserts 2 and 3 correspond to the CTD (C-terminal domain) of the S1 subunit in the 2019-nCoV spike glycoprotein. The insert 4 is at the junction of the SD1 (sub domain 1) and SD2 (sub domain 2) of the S1 subunit (Ou et al., 2017). We speculate, that these insertions provide additional flexibility to the glycoprotein binding site by forming a hydrophilic loop in the protein structure that may facilitate or enhance virus-host interactions.
bioRxiv preprint first posted online Jan. 31, 2020; doi: http://dx.doi.org/10.1101/2020.01.30.927871. The copyright holder for this preprint (which was not peer-reviewed) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC-ND 4.0 International license.
Figure 3. Modelled homo-trimer spike glycoprotein of 2019-nCoV virus. The inserts from HIV envelop protein are shown with colored beads, present at the binding site of the protein.
Evolutionary Analysis of 2019-nCoV
It has been speculated that 2019-nCoV is a variant of Coronavirus derived from an animal source which got transmitted to humans. Considering the change of specificity for host, we decided to study the sequences of spike glycoprotein (S protein) of the virus. S proteins are surface proteins that help the virus in host recognition and attachment. Thus, a change in these proteins can be reflected as a change of host specificity of the virus. To know the alterations in S protein gene of 2019-nCoV and its consequences in structural re-arrangements we performed in-sillico analysis of 2019-nCoV with respect to all other viruses. A multiple sequence alignment between the S protein amino acid sequences of 2019-nCoV, Bat-SARS-Like, SARS-GZ02 and MERS revealed that S protein has evolved with closest significant diversity from the SARS-GZ02 (Figure 1).
Insertions in Spike protein region of 2019-nCoV
Since the S protein of 2019-nCoV shares closest ancestry with SARS GZ02, the sequence coding for spike proteins of these two viruses were compared using MultiAlin software. We found four new insertions in the protein of 2019-nCoV- “GTNGTKR” (IS1), “HKNNKS” (IS2), “GDSSSG” (IS3) and “QTNSPRRA” (IS4) (Figure 2). To our surprise, these sequence insertions were not only absent in S protein of SARS but were also not observed in any other member of the Coronaviridae family (Supplementary figure). This is startling as it is quite unlikely for a virus to have acquired such unique insertions naturally in a short duration of time
bioRxiv preprint first posted online Jan. 31, 2020; doi: http://dx.doi.org/10.1101/2020.01.30.927871. The copyright holder for this preprint (which was not peer-reviewed) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC-ND 4.0 International license.
Insertions share similarity to HIV
The insertions were observed to be present in all the genomic sequences of 2019-nCoV virus available from the recent clinical isolates (Supplementary Figure 1). To know the source of these insertions in 2019-nCoV a local alignment was done with BLASTp using these insertions as query with all virus genome. Unexpectedly, all the insertions got aligned with Human immunodeficiency Virus-1 (HIV-1). Further analysis revealed that aligned sequences of HIV-1 with 2019-nCoV were derived from surface glycoprotein gp120 (amino acid sequence positions: 404-409, 462-467, 136- 150) and from Gag protein (366-384 amino acid) (Table 1). Gag protein of HIV is involved in host membrane binding, packaging of the virus and for the formation of virus-like particles. Gp120 plays crucial role in recognizing the host cell by binding to the primary receptor CD4.This binding induces structural rearrangements in GP120, creating a high affinity binding site for a chemokine co-receptor like CXCR4 and/or CCR5.
Discussion
The current outbreak of 2019-nCoV warrants a thorough investigation and understanding of its ability to infect human beings. Keeping in mind that there has been a clear change in the preference of host from previous coronaviruses to this virus, we studied the change in spike protein between 2019-nCoV and other viruses. We found four new insertions in the S protein of 2019-nCoV when compared to its nearest relative, SARS CoV. The genome sequence from the recent 28 clinical isolates showed that the sequence coding for these insertions are conserved amongst all these isolates. This indicates that these insertions have been preferably acquired by the 2019-nCoV, providing it with additional survival and infectivity advantage. Delving deeper we found that these insertions were similar to HIV-1. Our results highlight an astonishing relation between the gp120 and Gag protein of HIV, with 2019-nCoV spike glycoprotein. These proteins are critical for the viruses to identify and latch on to their host cells and for viral assembly (Beniac et al., 2006). Since surface proteins are responsible for host tropism, changes in these proteins imply a change in host specificity of the virus. According to reports from China, there has been a gain of host specificity in case 2019-nCoV as the virus was originally known to infect animals and not humans but after the mutations, it has gained tropism to humans as well.
Moving ahead, 3D modelling of the protein structure displayed that these insertions are present at the binding site of 2019-nCoV. Due to the presence of gp120 motifs in 2019-nCoV spike glycoprotein at its binding domain, we propose that these motif insertions could have provided an enhanced affinity towards host cell receptors. Further, this structural change might have also increased the range of host cells that 2019-nCoV can infect. To the best of our knowledge, the function of these motifs is still not clear in HIV and need to be explored. The exchange of genetic material among the viruses is well known and such critical exchange highlights the risk and the need to investigate the relations between seemingly unrelated virus families.
Conclusions
Our analysis of the spike glycoprotein of 2019-nCoV revealed several interesting findings: First, we identified 4 unique inserts in the 2019-nCoV spike glycoprotein that are not present in any other coronavirus reported till date. To our surprise, all the 4 inserts in the 2019-nCoV mapped to
bioRxiv preprint first posted online Jan. 31, 2020; doi: http://dx.doi.org/10.1101/2020.01.30.927871. The copyright holder for this preprint (which was not peer-reviewed) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC-ND 4.0 International license.
short segments of amino acids in the HIV-1 gp120 and Gag among all annotated virus proteins in the NCBI database. This uncanny similarity of novel inserts in the 2019- nCoV spike protein to HIV-1 gp120 and Gag is unlikely to be fortuitous. Further, 3D modelling suggests that atleast 3 of the unique inserts which are non-contiguous in the primary protein sequence of the 2019-nCoV spike glycoprotein converge to constitute the key components of the receptor binding site. Of note, all the 4 inserts have pI values of around 10 that may facilitate virus-host interactions. Taken together, our findings suggest unconventional evolution of 2019-nCoV that warrants further investigation. Our work highlights novel evolutionary aspects of the 2019-nCoV and has implications on the pathogenesis and diagnosis of this virus.
References
Beniac, D. R., Andonov, A., Grudeski, E., & Booth, T. F. (2006). Architecture of the SARS coronavirus prefusion spike. Nature Structural and Molecular Biology, 13(8), 751–752. https://doi.org/10.1038/nsmb1123
Biasini, M., Bienert, S., Waterhouse, A., Arnold, K., Studer, G., Schmidt, T., Kiefer, F., Cassarino, T. G., Bertoni, M., Bordoli, L., & Schwede, T. (2014). SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Research. https://doi.org/10.1093/nar/gku340
Bosch, B. J., van der Zee, R., de Haan, C. A. M., & Rottier, P. J. M. (2003). The Coronavirus Spike Protein Is a Class I Virus Fusion Protein: Structural and Functional Characterization of the Fusion Core Complex. Journal of Virology, 77(16), 8801–8811. https://doi.org/10.1128/jvi.77.16.8801- 8811.2003
Chan, J. F.-W., Kok, K.-H., Zhu, Z., Chu, H., To, K. K.-W., Yuan, S., & Yuen, K.-Y. (2020). Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerging Microbes & Infections, 9(1), 221–236. https://doi.org/10.1080/22221751.2020.1719902
Chan, J. F. W., Lau, S. K. P., To, K. K. W., Cheng, V. C. C., Woo, P. C. Y., & Yuen, K.-Y. (2015). Middle East Respiratory Syndrome Coronavirus: Another Zoonotic Betacoronavirus Causing SARS-Like Disease. https://doi.org/10.1128/CMR.00102-14
Chan, J., To, K., Tse, H., Jin, D., microbiology, K. Y.-T. in, & 2013, undefined. (n.d.). Interspecies transmission and emergence of novel viruses: lessons from bats and birds. Elsevier.
Corpet, F. (1988). Multiple sequence alignment with hierarchical clustering. Nucleic Acids Research. https://doi.org/10.1093/nar/16.22.10881
DeLano, W. L. (2002). The PyMOL Molecular Graphics System, Version 1.1. Schr{ö}dinger LLC. https://doi.org/10.1038/hr.2014.17
Du, L., Zhao, G., Kou, Z., Ma, C., Sun, S., Poon, V. K. M., Lu, L., Wang, L., Debnath, A. K., Zheng, B.-J., Zhou, Y., & Jiang, S. (2013). Identification of a Receptor-Binding Domain in the S Protein of the Novel Human Coronavirus Middle East Respiratory Syndrome Coronavirus as an Essential Target for Vaccine Development. Journal of Virology, 87(17), 9939–9942. https://doi.org/10.1128/jvi.01048- 13
bioRxiv preprint first posted online Jan. 31, 2020; doi: http://dx.doi.org/10.1101/2020.01.30.927871. The copyright holder for this preprint (which was not peer-reviewed) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC-ND 4.0 International license.
Edgar, R. C. (2004). MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research. https://doi.org/10.1093/nar/gkh340
Elbe, S., & Buckland-Merrett, G. (2017). Data, disease and diplomacy: GISAID’s innovative contribution to global health. Global Challenges. https://doi.org/10.1002/gch2.1018
Kirchdoerfer, R. N., Cottrell, C. A., Wang, N., Pallesen, J., Yassine, H. M., Turner, H. L., Corbett, K. S., Graham, B. S., McLellan, J. S., & Ward, A. B. (2016). Pre-fusion structure of a human coronavirus spike protein. Nature. https://doi.org/10.1038/nature17200
Kumar, S., Stecher, G., Li, M., Knyaz, C., & Tamura, K. (2018). MEGA X: Molecular evolutionary genetics analysis across computing platforms. Molecular Biology and Evolution. https://doi.org/10.1093/molbev/msy096
Li, F. (2016). Structure, Function, and Evolution of Coronavirus Spike Proteins. Annual Review of Virology, 3(1), 237–261. https://doi.org/10.1146/annurev-virology-110615-042301
Murakami, T. (2008). Roles of the interactions between Env and Gag proteins in the HIV-1 replication cycle. Microbiology and Immunology, 52(5), 287–295. https://doi.org/10.1111/j.1348- 0421.2008.00008.x
Ou, X., Guan, H., Qin, B., Mu, Z., Wojdyla, J. A., Wang, M., Dominguez, S. R., Qian, Z., & Cui, S. (2017). Crystal structure of the receptor binding domain of the spike glycoprotein of human betacoronavirus HKU1. Nature Communications. https://doi.org/10.1038/ncomms15216
Snijder, E. J., van der Meer, Y., Zevenhoven-Dobbe, J., Onderwater, J. J. M., van der Meulen, J., Koerten, H. K., & Mommaas, A. M. (2006). Ultrastructure and origin of membrane vesicles associated with the severe acute respiratory syndrome coronavirus replication complex. Journal of Virology, 80(12), 5927–5940. https://doi.org/10.1128/JVI.02501-05
Zhou, P., Yang, X.-L., Wang, X.-G., Hu, B., Zhang, L., Zhang, W., Si, H.-R., Zhu, Y., Li, B., Huang, C.-L., Chen, H.-D., Chen, J., Luo, Y., Guo, H., Jiang, R.-D., Liu, M.-Q., Chen, Y., Shen, X.-R., Wang, X., … Shi, Z.-L. (2020). Discovery of a novel coronavirus associated with the recent pneumonia outbreak in humans and its potential bat origin. BioRxiv. https://doi.org/10.1101/2020.01.22.914952
Zhu, N., Zhang, D., Wang, W., Li, X., Yang, B., Song, J., Zhao, X., Huang, B., Shi, W., Lu, R., Niu, P., Zhan, F., Ma, X., Wang, D., Xu, W., Wu, G., Gao, G. F., & Tan, W. (2020). A Novel Coronavirus from Patients with Pneumonia in China, 2019. New England Journal of Medicine, NEJMoa2001017. https://doi.org/10.1056/NEJMoa2001017
bioRxiv preprint first posted online Jan. 31, 2020; doi: http://dx.doi.org/10.1101/2020.01.30.927871. The copyright holder for this preprint (which was not peer-reviewed) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC-ND 4.0 International license.
Fig.S1 Multiple sequence alignment of glycoprotein of coronaviridae family, representing all the four inserts.
bioRxiv preprint first posted online Jan. 31, 2020; doi: http://dx.doi.org/10.1101/2020.01.30.927871. The copyright holder for this preprint (which was not peer-reviewed) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC-ND 4.0 International license.
Fig.S2: All four inserts are present in the aligned 28 Wuhan 2019-nCoV virus genomes obtained from GISAID. The gap in the Bat-SARS Like CoV in the last row shows that insert 1 and 4 is very unique to Wuhan 2019-nCoV.
bioRxiv preprint first posted online Jan. 31, 2020; doi: http://dx.doi.org/10.1101/2020.01.30.927871. The copyright holder for this preprint (which was not peer-reviewed) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC-ND 4.0 International license.
Fig.S3 Phylogenetic tree of 28 clinical isolates genome of 2019-nCoV including one from bat as a host.
bioRxiv preprint first posted online Jan. 31, 2020; doi: http://dx.doi.org/10.1101/2020.01.30.927871. The copyright holder for this preprint (which was not peer-reviewed) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC-ND 4.0 International license.
Supplementary Fig 4. Genome alingment of Coronaviridae family. Highlighted black sequences are the inserts represented here.
Decoding the evolution and transmissions of the novel pneumonia coronavirus (SARS-CoV-2) using whole genomic data
Wen-Bin Yu1,2 * Ph.D., Guang-Da Tang3,4 Ph.D., Li Zhang5 Ph.D., Richard T. Corlett1,2 Ph.D.
1Center for Integrative Conservation, Xishuangbanna Tropical Botanical Garden, Chinese Academy of Sciences, Mengla, Yunnan 666303, China
2Center of Conservation Biology, Core Botanical Gardens, Chinese Academy of Sciences, Mengla, Yunnan 666303, China
3Henry Fok College of Biology and Agriculture, Shaoguan University, Shaoguan 512005, China 4College of Forestry and Landscape Architecture, South China Agricultural University, Guangzhou 510642, China
5Chinese Institute for Brain Research, Beijing 102206, China
* Author for corresponding: yuwenbin@xtbg.ac.cn
1
chinaXiv:202002.00033v2
Summary
Background The outbreak of COVID-19 started in mid-December 2019 in Wuhan, Central China. Up to February 19, 2020, SARS-CoV-2 has infected more than 75,000 people in China, and another 25 countries across five continents. In this study, we used 93 complete genomes of SARS-CoV-2 from the GISAID EpiFluTM database to investigate the evolution and human-to-human transmissions of SARS-CoV-2 in the recent two months.
Methods Haplotype analyses were conducted on the alignment of coding regions using DnaSP. Evolutionary analysis of haplotypes used NETWORK. Population size changes were estimated using both DnaSP and Arlequin. Expansion date of population size was calculated based on the expansion parameter tau (τ) using the formula t=τ/2u.
Findings Eight coding-regions have 120 substitution sites, including 79 non-synonymous and 40 synonymous substitutions. Forty-two non-synonymous substitutions changed the biochemical property of amino acids. No combinations were detected. Fifty-eight haplotypes were classified into five groups: 31 haplotypes were found in samples from China and 31 in samples from other countries. The rooted network suggested that H13 and H35 were ancestral haplotypes, and H1 (which with its descendants included all samples from the Hua Nan market) was derived from the H3 haplotype. Population size of SARS-CoV-2 was estimated to have had a recent expansion on 6 January 2020, and an early expansion on 8 December 2019.
Interpretation Genomic variations of SARS-CoV-2 are still low in comparisons with published genomes of SARS-CoV and MERS-CoV. Phyloepidemiologic analyses suggested that the SARS-CoV-2 source at the Hua Nan market was imported from elsewhere. The crowded market then boosted SARS-CoV-2 circulation and spread it to the whole city in early December 2019. Furthermore, phyloepidemiologic approaches have recovered specific directions of human-to-human transmissions and the sources for international infected cases.
Funding Ten Thousand Talents Program of Yunnan, and Chinese Academy of Sciences.
2
chinaXiv:202002.00033v2
Evidence before this study
Up to February 19, 2020, SARS-CoV-2 had infected more than 75,000 people worldwide. Tracing back to the first identified COVID-19 patient on 1 December, SARS-CoV-2 has been circulated in humans for more than two months. However, it is still unclear whether the Hua Nan market was the birthplace of the virus, and how it has been transmitted and spread subsequently. We searched PubMed, preprint archives, and Google Scholar for articles published up to February 18, 2020, that contained information about the COVID-19 outbreak using the terms “2019-nCoV” or “SARS-CoV-2”, “coronavirus”, “evolution” or “phylogeny”, “GISAID”, “seafood market”, “transmission”, and “Wuhan”. We found six studies using fewer than 55 genomes of SARS-CoV-2 for phylogenetic analyses and molecular dating using BEAST.
Added value of this study
We found that 120 substitution sites were evenly distributed in eight coding regions, without evident recombination events. An earlier expansion date could be traced back to 8 December 2019. Genomic evidence did not support the Hua Nan market as the birthplace of SARS-CoV-2. In the first two months, most infected people were linked to Wuhan, but some infected patients outside China may link to Guangdong or other places.
Implications of all the available evidence
We suggest that SARS-CoV-2 may have already circulated widely among humans in Wuhan before December 2019, probably beginning in mid to late November. Some infected patients may have been overlooked because they had mild symptoms. We have demonstrated that a phylogenetic approach can be incorporated into epidemiological studies to search for the original source of SARS-CoV-2 and identify the direction of human-to-human transmissions.
3
chinaXiv:202002.00033v2
Introduction
Betacoronaviruses are characterized by enveloped, positive-sense, single-stranded RNA, and hosted in animals, particularly in mammals.1 Before December 2019, four species/strains of Betacoronavirus, HKU1, MERS-CoV, OC43 and SARS-CoV, had been reported to cause severe human diseases.1 The fifth species/strain, a novel betacoronavirus SARS-CoV-22 causing human pneumonia (i.e. COVID-19), was first reported in Wuhan, Hubei, Central China.3,4 Up to February 19, 2020, SARS-CoV-2 has infected more than 75,000 people in all provinces/regions of China, and another 25 countries across Africa, Asia, Europe, North America, and Oceania.5 Because SARS-CoV-2 can transmit from human to human,6 the massive exodus of people before the Chinese Spring Festival boosted the infection frequencies, as predicted.7 Daily confirmed infection cases were more than 2,000 between January 30 and 16 February 16, 2020, and the highest was more than 15,100,5 almost twice the total number of infection cases of SARS-CoV.8
As a member of subgenus Sarbecovirus, SARS-CoV-2 has been suggested to be of bat origin,3,9 and may have been transmitted to humans through non-bat intermediate mammals (e.g. pangolins10). Medical information for the first 41 infected patients in Wuhan showed that 27 patients were linked to the Hua Nan seafood market,6,11 which sold living wild mammals. This suggests a high possibility that SARS-CoV-2 originated in the market, then the infected people transmitted it to other people out of the market. However, this conclusion has been debated because the first identified infected person and 12 others had no link to the Hua Nan market. Some researchers have therefore argued that the Hua Nan market was not the original and/or only source of SARS-CoV-2 transmission to humans.12 The market was closed on 1 January 2020, making it very difficult to identify the intermediate animal vectors of SARS-CoV-2. In the absence of information on potential intermediary reservoirs, the origin and transmission pattern of SARS-CoV-2 are still unresolved.
Since the outbreak of COVID-19 was first identified in Wuhan in mid-December 2019, the first infected individuals identified in other provinces and regions of China, and other countries, during January 2020, have been assumed to have been infected in Wuhan or through contact with people from Wuhan.13-16 For example, the first identified infected patient and his four family members from Shenzhen visited Wuhan between Dec 29, 2019 and Jan 4, 2020,13 and the first identified infected patient in the United States visited family in Wuhan and returned to Washington State on January 15, 2020.14 SARS-CoV-2 can transmit from human to human,6 so Wuhan has been assumed to be the birthplace of SARS-CoV-2. However, this assumption has not been fully validated because the Hua Nan market has not been confirmed as the single source of SARS-CoV-2 transmission to humans and other possible original sources of SARS-CoV-2 have not been identified in Wuhan yet. It is evident, however, that the Hua Nan market boosted SARS-CoV-2 transmission to humans at an early stage of the pneumonia outbreak in Wuhan, after
which it spread rapidly with infected travelers to the whole of China and to other countries. 4
chinaXiv:202002.00033v2
In this study, we used 93 genomes of SARS-CoV-2 from the GISAID EpiFluTM database17 (access date 12 February 2020) to decode the evolution and transmissions of SARS-CoV-2 in the recent two months. Our aims were to: 1) characterize genomic variations of SARS-CoV-2; 2) infer the evolutionary relationships of the worldwide samples; and 3) deduce the transmission history of SARS-CoV-2 in Wuhan and out of Wuhan to the world.
Methods
To decode the evolutionary history of SARS-CoV-2, we retrieved 96 complete genomes from GISAID (Table S1, access by 12 February, 2020).17 The genome EPI_ISL_402131 (bat-RaTG13-CoV, hereafter) from GISAID was also included as the outgroup, because it is the closest sister betacoronavirus to SARS-CoV-2.3 The 97 genome sequences were aligned using MAFFT,18 then the alignment was manually checked using Geneious (Biomatters, New Zealand). In the alignment, we found that the genome EPI_ISL_404253 contains six ambiguous sites at variable positions and EPI_ISL_407079 and EPI_ISL_408978 have 175 “N” and 1,476 “N” bases, respectively, so these three genomes were excluded in this study. In addition, four genomes (EPI_ISL_407071, EPI_ISL_407894, EPI_ISL_407896, and EPI_ISL_409067) have their own private ambiguous sites, which were conservatively replaced by the common nucleotide at that position in the alignment. In the alignment, the 5’UTR and 3’UTR contain missing and ambiguous sites, so both regions were excluded in the following analyses.
The alignment was then imported into DnaSP19 for haplotype analyses. Population size changes were estimated based on a constant population size hypothesis using DnaSP, in combination with neutrality tests (Tajima’s D and Fu’s Fs). We also used Arlequin20 to test the sudden population expansion hypothesis and to calculate the expansion parameter tau (τ) if sudden population expansion is not rejected. We used the formula t=τ/2u to estimate the time since expansion (in days). In the formula, u is the cumulative substitution rate per year (for the genome sequence, so we used the formula u = μk to calculate it, where μ is the substitution rate per site per year, and k is the genome sequence length (29,358 bp for the CDS matrix). In this study, the substitution rate was set as 0·92×10-3 (95% CI, 0·33×10-3-1·46×10-3) substitution/site/year based on the most recent estimation for SARS-CoV-2.21 A median-joining network of haplotypes was generated by the NETWORK program22 with bat-RaTG13-CoV as the outgroup. Phylogenomic analyses of haplotypes were performed using IQ-TREE23. We conducted likelihood mapping and SH-like approximate likelihood ratio tests to assess the phylogenetic information and branch supports, respectively.
Results and discussion
Genomic variations of SARS-CoV-2
Genome size of SARS-CoV-2 varied from 29,782 bp to 29,903 bp. The aligned matrix was 29,910
5
chinaXiv:202002.00033v2
bp in length including 140 variable sites. The coding regions contained 120 substitution sites (Figure S1), which were classified as 58 haplotypes (Table S2). Nucleotide diversity (Pi) was 0·15×10-3±0·02×10-3 (Standard Deviation, SD, hereafter). Haplotype diversity (Hd) was 0·953±0·016 (SD) and variance of Hd was 0·26×10-3.
There are 120 substitution sites found in eight coding sequence (CDS) regions, i.e. Replicase polyprotein CDS (75 sites, 0·35% of the whole sequence), Spike glycoprotein CDS (16 sites, 0·42%), ORF3 (7 sites, 0·75%), Membrane glycoprotein CDS (4 sites, 0·60%), ORF7 (3 sites, 0·82%), ORF8 (3 sites, 0·82%), Nucleocapsid protein CDS (11 sites, 0·77%), and ORF10 (1 site, 0.86%), including 79 transitions (65·83%) and 41 transversions (34·17%). A chi-squared test showed that the distribution of substitution sites across ten CDS regions in the genome was even (χ2=1·958, df=9, P=0·99). Substitution sites at the 1st to 3rd frame positions are 27 (25·55%), 44 (40·0%), and 49 (44·55%), respectively. The 120 mutation sites are associated with 119 codons, including 79 non-synonymous (65·83%) and 40 synonymous (33·61%) substitutions. There were 42 non-synonymous substitutions (53·17%) which changed the biochemical properties of the amino acid (AA). The details for each CDS gene are shown in Figure 1 and Table S3. It is not clear whether non-synonymous substitutions and the biochemical property changes of amino acids might change the infectious activities of SARS-CoV-2. The current samplings showed that H1 haplotype has been found in 19 patients, but most haplotypes were just sequenced once. One possible explanation is that a common haplotype from the Hua Nan market (Figure 2, Table S1) was rapidly circulated at an early stage of human-to-human transmissions.
In comparisons with published genomes of SARS-CoV24 and MERS-CoV,25 genomic variations of SARS-CoV-2 are still low, without evident recombination sites/regions (Rm=2, P=1·0) at this time. According to the collection dates of the sequenced samples, haplotypes H1 and H3 were found in two samples at intervals of more than 30 days, and multiple samples over 20 days (Figure 2, Table S1). Although the incubation period can be over 24 days, there was only one case of this out of 1099 observations.26 Estimation of DNA substitution rates using 90 genomes of SARS-CoV-2 showed that the rate for SARS-CoV-2 (0·92×10-3, 95% CI: 0·33-1·46×10-3 substitution/site/year21) was lower than the rates for SARS-CoV (95% CI: 0·80-2·38×10-3 substitution/site/year27) and MERS-CoV (1·12×10-3, 95% CI: 0· ×10-3 substitution/site/year28; ×10-3, 95% CI: 0· ×10-3 substitution/site/year29). It therefore looks as if SARS-CoV-2 is still undergoing stable evolution. Due to mild symptoms and low mortality30,31, the immune system of the infected humans may provide a suitable environment for propagation of SARS-CoV-2. SARS-CoV-2 is highly infectious31 and is able to infect humans not only through the mucous membranes of the nose and mouth, but also use mucous membranes in the eyes,32 which may boost regional circulation and large-scale spread. Some large mutations may have occurred in Wuhan or other regions, but the strict quarantine policy over China since 23 January 2020 may have reduced the circulation and spreading of some mutants.
88-1·
37
or 0·
96
83-1·
09
6
chinaXiv:202002.00033v2
Of 93 genomes of SARS-CoV-2, 39 (41·93%) were from infected patients in 11 countries on four continents and encoded 31 haplotypes (Hd=0·987±0·009 (SD), Pi=0·16×10-3±0·01×10-3), with 27 nationally/regionally private haplotypes. The 54 genomes (58.07%) from China also encoded 31 haplotypes (Hd=0·906±0·001 (SD), Pi=0·14×10-3±0·03×10-3). A proportion Z-test showed significant differences in haplotype diversity of samples between China and other countries (χ2=4·024, df=1, P<0·05). The high haplotype diversity found in samples from other countries may be because the sampling dates were mostly after 22 January 2020, while those in China were before this date (Table S1 and Figure S2). In addition, the low level of radiation exposure on long-distance international flights33 may have accelerated mutation rates of SARS-CoV-2.34
Population size expansion of SARS-CoV-2
Constant population size of SARS-CoV-2 was rejected (Ramos-Onsins and Rozas’s R2=0·025, P<0·001; Raggedness r=0·011, P<0·05) using DnaSP (also see Figure S3), while both Fu’s test (Fs=-67.681.964, P<0·001) and Tajima’s D test (D=-2.701, P<0.001) indicated that the population size of SARS-CoV-2 was rapidly increasing. Mismatch distribution analysis using Arlequin strongly supported that the population of SARS-CoV-2 underwent sudden expansion (τ=2·887, Sum of Squared deviation=0·541×10-3, P=0·88, Harpending’s Raggedness index=0·010, P=0·88). The calculated expansion was 28·72 days (95% CI: 12·29-54·36 days) ago. Of the 93 genomes, the latest one was sampled on 3 February 2020, so the estimated expansion date was on 6 January 2020 (95% CI: 11 December 2019-22 January 2020), when China CDC started to activate a Level-2 emergency response.6 Until 6 January 2020, 129 patients were identified as SARS-CoV-2 infected through field investigations.6 Of 22 genomes (17·05 % of 129 patients) sequenced before 6 January 2020, 13 haplotypes (22.41% of 58 haplotypes) were recovered, which were H1 and its derived descendant haplotypes, and H3 (Figures 2 and 3). The CDS’s emergency response greatly reduced public activities and travels, and may have reduced the local circulation and large-scale spread in the following weeks of January.
Furthermore, mismatch distribution analysis of the 22 genomes before 6 January 2020 also showed a sudden population expansion of SARS-CoV-2 at an earlier stage of transmission (τ=2·818, Sum of Squared deviation=0·010, P=0·41, Harpending’s Raggedness index=0·046, P=0·57, Tajima’s D=-2·241, P<0·001; Fu’s Fs=-7·834, P<0·001). This earlier population expansion time was estimated at 28·38 days (95% CI: 12·00-54·36 days) before 5 January 2020, which was the latest sampling date of the 22 genomes. This earlier expansion was thus estimated to have occurred on 8 December 2019 (95% CI: 13 November 2019-26 December 2019), when there was only one infected patient officially reported.6,11 Therefore, SARS-CoV-2 had already circulated widely among humans in Wuhan before December 2019, probably beginning in mid to late November.21
7
chinaXiv:202002.00033v2
Evolutionary relationships of SARS-CoV-2 haplotypes
The evolutionary network of 58 haplotypes of SARS-CoV-2, with bat-RaTG13-CoV as the outgroup, is shown in Figure 3A. Five main groups can be recognized. In the network, H1, H3, and H13 were three core haplotypes, so Groups A-C were recognized using them as the central (i.e., ancestral super-spreader) haplotypes. Groups D and E were recognized based on two new super-spreader haplotypes, H56 and a medium vector mv2, which was a hypothesized (often ancestral) haplotype not sampled in the current samples. These two groups can be also treated as subgroups of Group C.
In the network, four satellite haplotypes and H35 connected to the H13 haplotype (Group A), and nine satellite haplotypes and H38+H45 and H50 haplotypes connected to H3 (Group B). The evolutionary network showed that bat-RaTG13-CoV connected through a hypothesized haplotype (mv1) to the H13 and H38 haplotypes by single mutations at positions 18,067 (S, synonymous substitution) and 29,102 (S), referring to the alignment length 29,910 bp. The connections between the H3 and H1 haplotypes are two mutations at positions 8,789 (S) and 28,151 (Ns, non-synonymous substitution). The H1 haplotype, the most abundant, included 19 samples, while 26 satellite haplotypes and H40+(H43 and H47) haplotypes are directly derived from the H1 haplotype (Group C). Moreover, five haplotypes of Group D and four haplotypes of Group E should be also derived from the H1 haplotype.
Where are the original sources from?
The evolutionary network suggests that the hypothesized haplotype mv1 may be from an intermediate host or the first infected humans. From those connections, both H13 and H38 would be suggested as ancestral haplotypes. The SH-like approximate likelihood ratio test showed both haplotype H13’s group and H38 (with H45) could be the most basal clades in phylogenies of the 58 haplotypes (Figure S4), but phylogenetic information of the alignment was informative (Figure S5). Two main evolutionary paths of available haplotypes can be from H13 through H3 to H1, or from H38 through H3 to H1 (Figure 3C). Both scenarios demonstrate H3 was the key connection from an ancestral haplotype to H1. Neither H13 nor H38 has samples from Wuhan (Hubei) (Figure 3). H13 was only recovered from five Shenzhen (Guangdong) samples, including the father (patient 2) of the familial cluster, who was one of the first identified infected patients in Guangdong.13 Two derived haplotypes were also only found in Shenzhen (H14 from the grandson of patient 2), and the other three haplotypes were found in three samples from Japan and one sample from Arizona in the United States (Figure 3). According to an epidemiological study, the Shenzhen family traveled to Wuhan after the outbreak was announced, and they could have been infected during their visit in Wuhan from a hospital or an unknown common source13. This suggests that H13 should have originated from Wuhan, but none of the available samples from Wuhan encode haplotypes in Group A. Genetically, haplotypes of Group A have links to only
Wuhan haplotype H3 (only one sample EPI_ISL_406801, with no link to the Hua Nan market). It 8
chinaXiv:202002.00033v2
is possible that H13 was newly derived from H3 in the family from Shenzhen (Figure 3C) and did not spread in Wuhan, or that no samples have been sequenced yet. However, this scenario is not supported by the evolutionary network. H38 has three genomes from the same patient (Table S1), who was the first identified infected patient in the United States.14 He should have been infected while visiting his family in China. The original source of H38 can be explained as that of H13, which is also derived from H3, and the derived H45 was from a Chongqing patient who was working in Wuhan.
The H3 haplotype has only one sample from Wuhan, which was not linked to the Hua Nan market,9 and the other samples in this group were from other countries and regions (Figure 3). Noteworthily, all the samples from the Hua Nan market had the H1 haplotype or its derived haplotypes (H2, H8-H12, see Figure 2 and Table S1), indicating that there were circulated infections within the market in the short term. It is possible that SARS-CoV-2 in the Hua Nan market had been transmitted from other places (Figure 3D), or at least, that Hua Nan market did not host the original source of SARS-CoV-2. As the first identified infected patients had no link to the market,11 it is possible that infected humans transmitted SARS-CoV-2 to workers or sellers in the market, after which it rapidly circulated there. The crowded market boosted SARS-CoV-2 transmissions to buyers and spread it to the whole city in early December 2019, corresponding to the estimated population expansion time.
Regional and worldwide circulation and spread
Of the 54 genomes from patients in China, Chongqing (3 samples), Guangdong (18), Hubei (22), Taiwan (2), and Zhejiang (4) have more than two samples, and the other five provinces sequenced one sample. Hubei (Wuhan) samples from 24 December 2019 to 5 January 2020 encoded 13 haplotypes, belonging to Groups C (H1 and 11 satellite haplotypes) and B (only H3). These relationships indicate a rapid transmission and circulations of SARS-CoV-2 in Wuhan at an early stage of transmission. H1 (no satellite haplotypes) and H3 haplotypes are the ancestors of haplotypes out of Wuhan/Hubei. Eighteen Guangdong samples, collected from 10 to 23 January 2020, encoded 15 haplotypes, belonging to Groups A, C and E, showing that there were multiple sources imported into Guangdong. Three haplotypes (H14, H15 and H17) may have evolved locally, indicating that human-to-human transmissions happened when SARS-CoV-2 initially spread to Shenzhen of Guangdong.13 Two samples from Taiwan encoded H3 and H24 in Groups B and D, respectively, and three samples from Chongqing encoded H1, H40, and H45 in Groups B and C, respectively. There were two sources imported into these two provinces/regions. Four Zhejiang samples encoded H1 and H24 in Group C, which was only imported from the source of the H1 haplotype.
The samples outside of China encoded 31 haplotypes belonging to Groups A-E. Of these, 27
haplotypes are private by regional samplings, only two Thailand samples were the H1 haplotype, 9
chinaXiv:202002.00033v2
one each from Australia and Belgium were the H3 haplotype, one sample from the United States was the H19 haplotype, and one sample from Singapore was the H40 haplotype. Twelve samples, encoding 10 haplotypes, were from patients in five countries in Asia. Seven haplotypes linked to Wuhan and three haplotypes linked to Guangdong (Shenzhen). Human-to-human transmission may have happened from patients with H53 to H52 haplotypes in Tokyo, Japan, who were repatriated Japanese from Wuhan.35 Five Oceanian samples, encoding six haplotypes in Groups B, C, and D, were from patients from three states in Australia, all with links to Wuhan. Patients with H3, with H25 and H26, and with H55 (linked to H1) were directly from Wuhan, and human-to-human transmission was from patients of H25 to H26, who were in a same tour group in Queensland.36 The connection between patients with H56 and H27 is not clear, because the patient with H56 flew to Sydney from Wuhan on 25 January 2020, and the patient with H27 flew to Melbourne from Wuhan on 15 January 2020. One possibility is that there was an intermediary spreader, who also transmitted SARS-CoV-2 to other patients in France, the United States, and Taiwan. Eight European samples, encoding seven haplotypes, were from patients in four countries. Patients in Belgium37 and Germany16 traveled to or stayed in Wuhan. Patients in England did not report a link to Wuhan,38 but a familial transmission was recovered from H28 to H29. Patients in France may have been infected by three different sources, i.e. H44 linked to Wuhan, H43 may link to Chongqing/Singapore, and H30 may link to an intermediary spreader.
Of the 13 genomes from the United States, three were from the same patient in Washington encoding the same haplotype H38, while the other three samples encoded eight haplotypes, covering all five groups (Figure 3A), so the sources of imported infections are complicated. Three haplotypes (H1 (California), H19 (Wisconsin), and H38 (Washington)) were linked to Wuhan, and three (H19 (Wisconsin), H35 (Arizona), H42 (California)) to five (H41 (California) and H58 (Illinois)) haplotypes may link to Guangdong. The remaining haplotypes (H36 (California), H37 (California), and H57 (Massachusetts)) linked to patients out of China (H54 (Vietnam) and H56 (Australia)), who were from Wuhan.15,39 It is not clear where they were infected. There is no human-to-human transmission evidence in the United States from the 11 cases.
Phylogenetic approaches provide insight into the epidemiology of SARS-CoV-2
Epidemiological study of SARS-CoV-2 using traditional approaches is very difficult, because it was not identified as a new coronavirus until 29 December, and some infected people with mild symptoms or without symptoms16,40 may have been overlooked in late November and early December. Moreover, the Hua Nan market, which was considered as the birthplace of SARS-CoV-2, has been closed since 1 January 2020. In this study, we have used genomic data for SARS-CoV-2 to infer evolutionary relationships of the 58 haplotypes, and to suggest that H1 and its descendant haplotypes from the Hua Nan market should be derived from the H3 haplotype, which was not linked to the market (Figure 3C). This suggests that the source of the coronavirus in
the Hua Nan market was imported from elsewhere, as also suggested by other researchers.12 The 10
chinaXiv:202002.00033v2
phylogenetic network indicated that H13 and H38 should be ancestral haplotypes that connected to the outgroup bat-RaTG13-CoV through a hypothesized intermediate haplotype. Because the currently available samples do not include the first identified infected patient and other patients from early December, the most common ancestral haplotype might be missed. If there are any frozen samples from those patients, it would be worth doing genomic sequencing for phyloepidemiologic study to help to locate the birthplace of SARS-CoV-2 in Wuhan. Meanwhile, we expect that the H13 and H38 haplotypes will be found in some samples of infected patients in Wuhan if more samples are sequenced in future. This will be very important in the search for the original sources of SARS-CoV-2, because both H13 and H38 tend to be ancestral haplotypes.
The evolutionary network of haplotypes can be used to recover the directions of human-to-human transmissions at the local scale and spread at the larger scale. The central haplotype can be considered as the super-spreader haplotype, and the tip haplotype is the most recent descendant haplotype. The transmission direction can be identified using the connection information of tips and branches. For example, the confirmed patients from the Hua Nan market shared the common ancestral haplotype H1, indicating they were infected from a common source, who may have been a super-spreader in the market. This transmission phenomenon may also have happened in Shenzhen with the patients of Group A. This approach has recovered specific directions of human-to-human transmission in the Shenzhen family (H13 → H14), the Queensland tour group (H25 → H26), the England family (H28 → H29), and the repatriated Japanese from Wuhan (H53 → H52). Most international infections link to Wuhan directly or indirectly, but for some of them it is not clear exactly where they were infected. As discussed above, we have found that some patients in Japan and United States might have been infected in Guangzhou, and one patient in France might have been infected in Chongqing or Singapore. We suspect that super-spreaders mediate the spreading from China to worldwide. At least, the infected people with H56 and mv2, as well as H54, contributed at least three haplotypes (Figure 3A).
Contributors
W-BY conceived the research, analyzed the data, interpreted the results, and wrote the draft manuscript; WBY and GDT collected data; all authors reviewed and approved the final version of the manuscript.
Declaration of interests
We declare no competing interests.
Acknowledgements
We are grateful to scientists and researchers for depositing whole genomic sequences of Novel Pneumonia Coronavirus (SARS-CoV-2) at the Global Initiative on Sharing All Influenza Data (GISAID) EpiFluTM; to GISAID database for allowing us to access the sequences for
11
chinaXiv:202002.00033v2
non-commercial scientific research; and to Dr. Jin Chen and Dr. De-Zhu Li for their valuable comments and suggestions on conceiving the research and an early version of the manuscript. WBY also thanks his wife Dr. Nan Jiang for supporting him in daily life during the quarantine of the Novel Pneumonia outbreak, so he can focus on this research. This study was supported by grants from the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB31000000), Ten Thousand Talents Program of Yunnan for Top‐notch Young Talents, and the open research project of “Cross-Cooperative Team” of the Germplasm Bank of Wild Species, Kunming Institute of Botany, Chinese Academy of Sciences.
References
1. Cui J, Li F, Shi Z-L. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol 2019; 17(3): 181-92.
2. Gorbalenya AE, Baker SC, Baric RS, et al. Severe acute respiratory syndrome-related coronavirus: The species and its viruses – a statement of the Coronavirus Study Group. 2020: 2020.02.07.937862.
3. Zhou P, Yang X-L, Wang X-G, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020; doi: 10.1038/s41586-020-2012-7.
4. Zhu N, Zhang D, Wang W, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med 2020: 10.1056/NEJMoa2001017.
5. A live infection database of the 2019-nCoV outbreak 2020. http://fms.news.cn/swf/2020_sjxw/2_1_xgyq/index.html (accessed Feb 8 2020).
6. Li Q, Guan X, Wu P, et al. Early transmission dynamics in Wuhan, China, of novel coronavirus–infected pneumonia. N Engl J Med 2020: 10.1056/NEJMoa2001316.
7. Wu JT, Leung K, Leung GM. Nowcasting and forecasting the potential domestic and international spread of the 2019-nCoV outbreak originating in Wuhan, China: a modelling study. Lancet 2020: 10.1016/S0140-6736(20)30260-9.
Chan-Yeung M, Xu R-H. SARS: epidemiology. Respirology 2003; 8(s1): S9-S14.
Lu R, Zhao X, Li J, et al. Genomic characterisation and epidemiology of 2019 novel
coronavirus: implications for virus origins and receptor binding. Lancet 2020: 10.1016/S0140-6736(20)30251-8.
10. Lam TT-Y, Shum MH-H, Zhu H-C, et al. Identification of 2019-nCoV related coronaviruses in Malayan pangolins in southern China. 2020: 2020.02.13.945485.
11. Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020: 10.1016/S0140-6736(20)30183-5.
12. Cohen J. Wuhan seafood market may not be source of novel virus spreading globally. Science 2020: 10.1126/science.abb0611.
13. Chan JF-W, Yuan S, Kok K-H, et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet 2020: 10.1016/S0140-6736(20)30154-9.
12
chinaXiv:202002.00033v2
14. Holshue ML, DeBolt C, Lindquist S, et al. First Case of 2019 Novel Coronavirus in the United States. N Engl J Med 2020: 10.1056/NEJMoa2001191.
15. Phan LT, Nguyen TV, Luong QC, et al. Importation and human-to-human transmission of a novel coronavirus in Vietnam. N Engl J Med 2020: 10.1056/NEJMc2001272.
16. Rothe C, Schunk M, Sothmann P, et al. Transmission of 2019-nCoV infection from an asymptomatic contact in Germany. N Engl J Med 2020: 10.1056/NEJMc2001468.
17. Shu Y, McCauley J. GISAID: Global initiative on sharing all influenza data – from vision to reality. Euro Surveill 2017; 22(13): 30494.
18. Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 2013; 30(4): 772-80.
19. Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC, et al. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol 2017; 34(12): 3299-302.
20. Excoffier L, Lischer HEL. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 2010; 10(3): 564-7.
21. Rambaut A. Phylodynamic analysis | 90 genomes | 12 Feb 2020. 2020. http://virological.org/t/phylodynamic-analysis-90-genomes-12-feb-2020/356 (accessed Feb 12 2020).
22. Bandelt HJ, Forster P, Röhl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 1999; 16(1): 37-48.
23. Minh BQ, Schmidt HA, Chernomor O, et al. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol 2020.
24. Luk HKH, Li X, Fung J, Lau SKP, Woo PCY. Molecular epidemiology, evolution and phylogeny of SARS coronavirus. Infect Genet Evol 2019; 71: 21-30.
25. Cotten M, Watson SJ, Kellam P, et al. Transmission and evolution of the Middle East respiratory syndrome coronavirus in Saudi Arabia: a descriptive genomic study. Lancet 2013; 382(9909): 1993-2002.
26. Guan W-j, Ni Z-y, Hu Y, et al. Clinical characteristics of 2019 novel coronavirus infection in China. medRxiv 2020: 2020.02.06.20020974.
27. Zhao Z, Li H, Wu X, et al. Moderate mutation rate in the SARS coronavirus genome and its implications. BMC Evol Biol 2004; 4: 21.
28. Cotten M, Watson SJ, Zumla AI, et al. Spread, circulation, and evolution of the middle rast tespiratory syndrome Coronavirus. mBio 2014; 5(1): e01062-13.
29. Dudas G, Carvalho LM, Rambaut A, Bedford T. MERS-CoV spillover at the camel-human interface. 2018; 7: e31257.
30. Zhang R, Liu H, Li F, et al. Transmission and epidemiological characteristics of Novel Coronavirus (2019-nCoV) Pneumonia (NCP): preliminary evidence obtained in comparison with 2003-SARS. medRxiv 2020: 2020.01.30.20019836.
31. Yang Y, Lu Q, Liu M, et al. Epidemiological and clinical features of the 2019 novel
coronavirus outbreak in China. medRxiv 2020: 2020.02.10.20021675. 13
chinaXiv:202002.00033v2
32. Lu C-w, Liu X-f, Jia Z-f. 2019-nCoV transmission through the ocular surface must not be ignored. Lancet 2020: 10.1016/S0140-6736(20)30313-5.
33. Bottollier-Depois J-F, Chau Q, Bouisset P, Kerlau G, Plawinski L, Lebaron-Jacobs L. Assessing exposure to cosmic radiation during long-haul flights. Radiat Res 2000; 153(5): 526-32, 7.
34. Shibai A, Takahashi Y, Ishizawa Y, et al. Mutation accumulation under UV radiation in Escherichia coli. Sci Rep 2017; 7(1): 14531.
35. Japan tightens immigration as 3 more infected by coronavirus. 2020. http://www.asahi.com/ajw/articles/AJ202002020013.html (accessed 2 February 2020).
36. Coronavirus outbreak: Second case confirmed in Queensland. 2020. https://7news.com.au/lifestyle/health-wellbeing/qld-coronavirus-case-remains-in-isolation-c-6715 00.
37. Belgium: First case of coronavirus confirmed in Belgium February 3. 2020. https://www.garda.com/fr/crisis24/alertes-de-securite/311116/belgium-first-case-of-coronavirus-co nfirmed-in-belgium-february-3 (accessed 4 February 2020).
38. UK confirms first new coronavirus case; rises risk level. 2020. https://www.pharmaceutical-technology.com/news/uk-coronavirus-case/ (accessed 31 January 2020).
39. Fifth Australian coronavirus case confirmed as 21-year-old UNSW student. 2020. https://www.sbs.com.au/news/fifth-australian-coronavirus-case-confirmed-as-21-year-old-unsw-st udent (accessed 27 January 2020).
40. Heymann DL, Shindo N. COVID-19: what is next for public health? Lancet 2020: 10.1016/S0140-6736(20)30374-3.